
Alcohol Metabolism and the ALDH2 Variant: The Biological Basis for Intolerance and DNA Damage
Overview
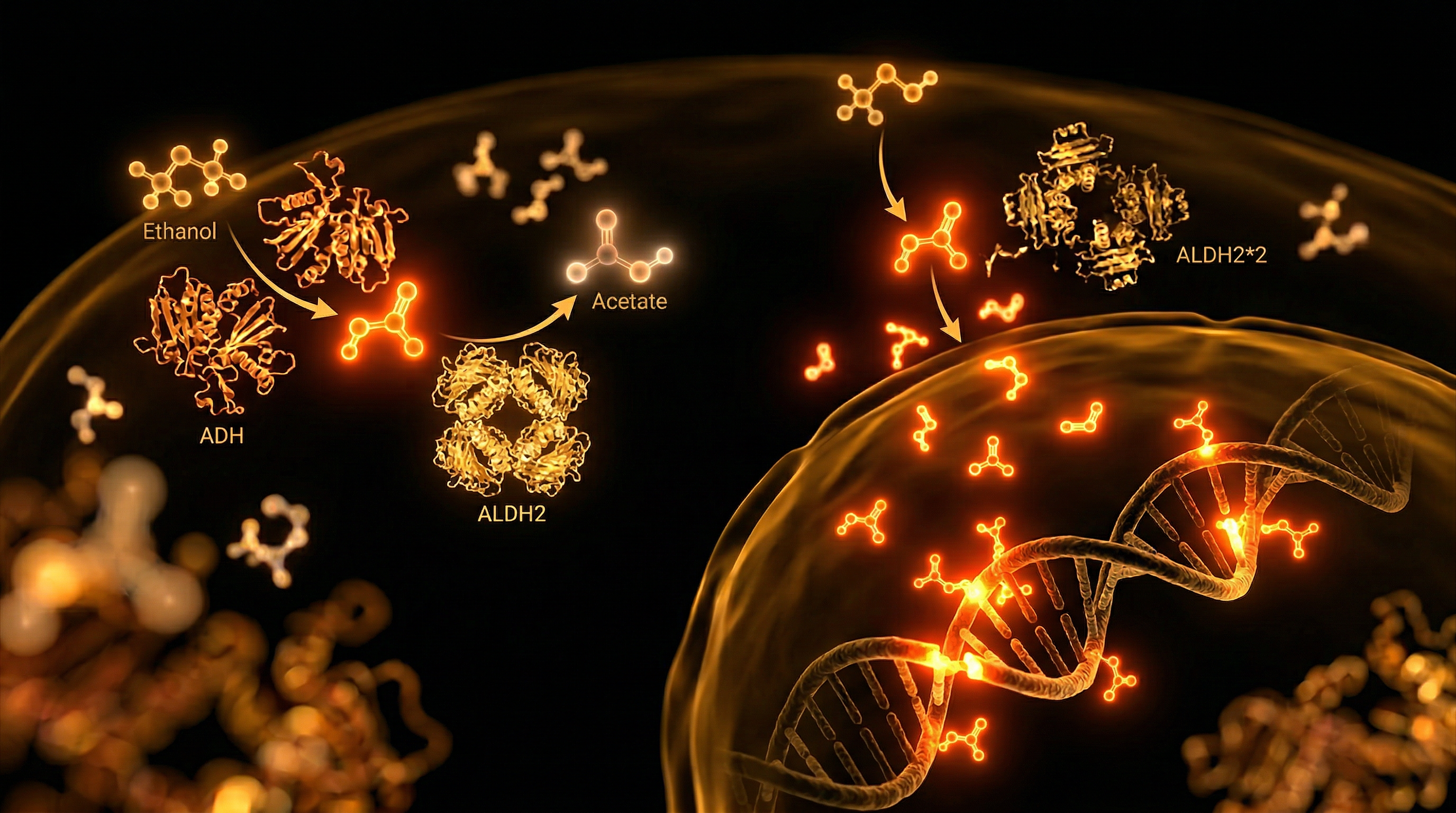
The detoxification of ethanol represents a rigorous two-stage oxidative challenge for the human hepatic system, governed primarily by the coordinated action of alcohol dehydrogenase (ADH) and aldehyde dehydrogenase 2 (ALDH2). While the conversion of ethanol to acetaldehyde via ADH is relatively efficient, the subsequent oxidation of acetaldehyde into inert acetate by the mitochondrial enzyme ALDH2 constitutes the critical rate-limiting step in systemic clearing. In a significant proportion of the global population, this metabolic bottleneck is exacerbated by the ALDH2*2 polymorphism (rs671), a single nucleotide polymorphism (SNP) characterised by a guanine-to-adenine transition. This genetic variant results in the substitution of glutamate with lysine at position 504 (Glu504Lys), a structural alteration that catastrophically diminishes the enzyme's catalytic affinity for its substrate. For individuals carrying the homozygous (AA) or even the heterozygous (GA) genotype, the result is a near-total or significant loss of ALDH2 activity, leading to the systemic accumulation of acetaldehyde—a highly reactive, electrophilic, and DNA-damaging intermediate.
At INNERSTANDIN, we must look beyond the superficial clinical manifestation of the "alcohol flushing response" to the more insidious molecular reality: acetaldehyde is classified by the International Agency for Research on Cancer (IARC) as a Group 1 carcinogen. When ALDH2 function is compromised, acetaldehyde persists in the bloodstream and tissues, where it exerts potent mutagenic effects. It readily reacts with DNA to form stable covalent adducts, most notably $N^2$-ethyl-2'-deoxyguanosine ($N^2$-Et-dG). These adducts disrupt base pairing and induce genomic instability, significantly elevating the risk of squamous cell carcinomas of the oesophagus and upper aerodigestive tract. Furthermore, the persistence of acetaldehyde triggers a cascade of oxidative stress; it promotes the production of reactive oxygen species (ROS), depletes cellular glutathione reserves, and induces lipid peroxidation, particularly within the mitochondria.
In the United Kingdom, where alcohol consumption is deeply integrated into social frameworks, the implications of ALDH2 deficiency are often overlooked by traditional medical assessments. Peer-reviewed evidence published in *The Lancet* and *Nature* suggests that the physiological burden of this variant extends far beyond intolerance, impacting cardiovascular health through increased systemic inflammation and arterial stiffness. The ALDH2*2 variant effectively removes the primary biological shield against alcohol-derived toxicity, transforming a common lifestyle choice into a primary driver of chromosomal aberrations and accelerated cellular senescence. This section explores the structural biology of the ALDH2 enzyme and the precise mechanisms by which metabolic failure translates into irreversible DNA cross-linking and systemic pathology, exposing the objective biological truth behind alcohol-induced genomic damage.
The Biology — How It Works
The oxidative metabolism of ethanol is not a singular event but a precarious two-step enzymatic relay that, when disrupted by genetic variation, transforms a common social lubricant into a potent endogenous mutagen. At the heart of this process lies the conversion of ethanol to acetaldehyde via alcohol dehydrogenase (ADH), followed by the rapid oxidation of acetaldehyde into inert acetate by the mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2). While ethanol itself possesses neurotoxic properties, it is the intermediate metabolite, acetaldehyde, that poses the most significant threat to cellular integrity. Classified as a Group 1 carcinogen by the International Agency for Research on Cancer (IARC), acetaldehyde is a highly reactive electrophile capable of inducing profound structural damage to DNA and proteins.
For individuals carrying the ALDH2*2 polymorphism (specifically the rs671 single nucleotide polymorphism), this metabolic relay suffers a catastrophic failure. This variant involves a G-to-A transition in exon 12 of the ALDH2 gene, resulting in a glutamate-to-lysine substitution at position 504 (Glu504Lys). At the molecular level, this substitution occurs near the enzyme's active site and its interaction domain with the NAD+ co-factor. Because ALDH2 functions as a tetramer, the presence of even a single mutant subunit can drastically reduce the catalytic efficiency of the entire complex. In heterozygotes (ALDH2*1/*2), enzymatic activity is reduced to approximately 10–15% of normal levels, while homozygotes (ALDH2*2/*2) possess near-zero functional capacity to clear acetaldehyde. This creates a systemic "acetaldehyde storm" where blood concentrations of the toxin can reach levels 20 times higher than those observed in individuals with the wild-type genotype.
The biological consequence of this accumulation extends far beyond the immediate "flush" response. Acetaldehyde’s primary mechanism of toxicity is its ability to form covalent DNA adducts. It reacts with deoxyguanosine to form $N^2$-ethylidene-2'-deoxyguanosine ($N^2$-ethylidene-dG), which can then be reduced to more stable $N^2$-ethyl-2'-deoxyguanosine ($N^2$-Et-dG) adducts. Research published in *Nature* and *The Lancet Oncology* has demonstrated that these adducts trigger DNA-protein crosslinks and double-strand breaks. In the absence of efficient clearance, these lesions overwhelm the Fanconi anaemia (FA) DNA repair pathway, leading to chromosomal instability and the permanent alteration of haematopoietic stem cells.
At INNERSTANDIN, we recognise that this is not merely an "intolerance" but a fundamental genomic vulnerability. In the UK context, where alcohol consumption is a major driver of public health crises, the presence of the ALDH2*2 variant—though most prevalent in East Asian populations—serves as a critical model for understanding alcohol-induced oncogenesis across all demographics. The inability to neutralise acetaldehyde forces the body to rely on secondary, less efficient pathways like CYP2E1, which simultaneously generates reactive oxygen species (ROS), further compounding mitochondrial distress and lipid peroxidation. This synergistic assault on the genome underscores why ALDH2 deficiency is one of the most significant genetic determinants of oesophageal squamous cell carcinoma and systemic multi-organ damage.
Mechanisms at the Cellular Level
At the crux of alcohol intolerance lies a profound enzymatic failure within the mitochondrial matrix, specifically involving the mitochondrial aldehyde dehydrogenase 2 (ALDH2) enzyme. In the standard metabolic pathway, ethanol is oxidised to acetaldehyde by alcohol dehydrogenase (ADH), a process followed immediately by the rapid conversion of acetaldehyde into inert acetate by ALDH2. However, for individuals carrying the ALDH2*2 polymorphism (rs671)—a single nucleotide polymorphism (SNP) characterised by a G-to-A transition resulting in a glutamate-to-lysine substitution at position 504 (Glu504Lys)—this process is catastrophically stalled. This variant acts in a dominant-negative fashion; because the functional ALDH2 enzyme is a tetramer, the presence of even a single mutant subunit drastically reduces the catalytic efficiency of the entire complex, leading to a near-total loss of enzymatic activity in homozygotes and a 60–80% reduction in heterozygotes.
The consequence of this bottleneck is the systemic accumulation of acetaldehyde, a highly reactive and electrophilic Group 1 carcinogen. At the cellular level, acetaldehyde exerts its most deleterious effects by forming covalent DNA adducts. The primary lesion, N2-ethyl-2'-deoxyguanosine (N2-Et-dG), disrupts base pairing and, if not excised, can lead to the formation of interstrand cross-links (ICLs). These ICLs are particularly lethal as they physical block replication forks and transcription machinery. Research from the MRC Laboratory of Molecular Biology in Cambridge has demonstrated that when the ALDH2-mediated clearance mechanism is absent, the body must rely heavily on the Fanconi Anaemia (FA) DNA repair pathway to resolve these cross-links. In the absence of efficient repair, acetaldehyde-induced damage leads to double-strand breaks, chromosomal instability, and the progressive depletion of haematopoietic stem cells, providing a direct mechanistic link between alcohol metabolism and bone marrow failure or leukaemogenesis.
Beyond direct genotoxicity, the ALDH2*2 variant induces a state of chronic oxidative stress and metabolic dysfunction. The failure to clear acetaldehyde promotes the production of reactive oxygen species (ROS) through the overactivation of NADPH oxidase and mitochondrial respiratory chain interference. This oxidative environment further depletes the pool of reduced glutathione (GSH), the cell's primary antioxidant, and triggers lipid peroxidation, resulting in the formation of secondary adducts like malondialdehyde (MDA). Furthermore, the altered NADH/NAD+ ratio caused by inefficient acetaldehyde processing disrupts the methionine cycle and cellular methylation potential. High levels of NADH inhibit NAD+-dependent enzymes such as the sirtuin family (SIRT1), which are critical for epigenetic regulation and DNA repair signalling. This intersection of SNP-driven metabolic failure and epigenetic dysregulation is a central pillar of the INNERSTANDIN methodology, exposing how a single genetic variant can cascade into systemic genomic instability and accelerated cellular senescence. For the UK population, where diverse genetic lineages intersect, identifying these SNP-driven metabolic bottlenecks is essential for quantifying individual risk profiles for oesophageal and upper aerodigestive tract cancers, where the synergy between ALDH2 deficiency and acetaldehyde exposure is most predatory.
Environmental Threats and Biological Disruptors
The metabolic degradation of ethanol is frequently mischaracterised as a linear clearance process; in reality, for those carrying the ALDH2*2 polymorphism (rs671), it represents a catastrophic failure of endogenous detoxification. At the heart of this disruption is the accumulation of acetaldehyde, a highly reactive electrophile and a Group 1 carcinogen as classified by the International Agency for Research on Cancer (IARC). While the wild-type ALDH2 enzyme efficiently oxidises acetaldehyde into inert acetate, the Glu504Lys substitution results in a near-total loss of catalytic activity. For the individual, this isn't merely a "flush reaction"—it is a systemic assault on genomic integrity and cellular homeostasis.
Acetaldehyde functions as a potent biological disruptor by inducing DNA-protein crosslinks (DPCs) and interstrand crosslinks (ICLs), which physically obstruct the replication machinery. Research published in *Nature* and *The Lancet* underscores that in the absence of functional ALDH2, the body relies heavily on the Fanconi Anaemia (FA) DNA repair pathway. When this second line of defence is saturated by chronic ethanol exposure or environmental aldehydes, the result is double-strand breaks and permanent chromosomal rearrangements. This is particularly relevant in the UK context, where the intersection of high alcohol consumption and the prevalence of ALDH2 variants in specific sub-populations—notably those of East Asian descent—creates a silent epidemic of oesophageal and upper aerodigestive tract cancers.
Furthermore, the threat extends beyond exogenous ethanol. The ALDH2 enzyme is the primary safeguard against endogenous aldehydes generated via lipid peroxidation and the metabolism of biogenic amines. Environmental stressors ubiquitous in British urban centres, such as formaldehyde from building materials and acrolein from vehicular emissions, synergise with ALDH2 deficiency. These "environmental disruptors" compete for the limited enzymatic bandwidth of a compromised ALDH2 system, leading to a pro-inflammatory state characterised by the elevation of 4-hydroxy-2-nonenal (4-HNE) adducts.
Crucially, from the perspective of INNERSTANDIN, we must address the epigenetic fallout. Elevated acetaldehyde levels exert a profound inhibitory effect on DNA methyltransferases (DNMTs). This disruption of the methyl cycle leads to global DNA hypomethylation and the aberrant silencing of tumour suppressor genes. The sequestration of S-adenosylmethionine (SAMe) to combat oxidative stress further starves the methylation pathway, creating a vicious cycle of genomic instability. This is not merely an intolerance; it is a fundamental breakdown of the biological barriers intended to protect the human blueprint from chemical degradation. The ALDH2 variant, therefore, serves as a magnifying glass for environmental toxicity, rendering the individual hyper-susceptible to insults that a wild-type metabolism might otherwise neutralise.
The Cascade: From Exposure to Disease
The ingestion of ethanol initiates a metabolic sequence that, in the presence of the ALDH2*2 polymorphism, shifts from a routine detoxification process to a systemic pathological cascade. Under normotypic conditions, ethanol is oxidised to acetaldehyde by alcohol dehydrogenase (ADH), which is then rapidly converted to inert acetate by the mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2). However, for individuals carrying the rs671 single nucleotide polymorphism (SNP)—a substitution of glutamate with lysine at position 504 (Glu504Lys)—the catalytic activity of the ALDH2 tetramer is virtually abolished. This enzymatic bottleneck results in an immediate and profound accumulation of acetaldehyde, a highly reactive electrophile and a confirmed Group 1 carcinogen according to the International Agency for Research on Cancer (IARC).
The biological consequences of this accumulation extend far beyond the transient cutaneous flushing often observed. At a molecular level, acetaldehyde exerts its toxicity by forming covalent DNA adducts, most notably N2-ethylidene-2'-deoxyguanosine (N2-ethyl-dG). Research published in *Nature* (Garaycoechea et al., 2018) has elucidated that when the ALDH2-mediated clearance pathway fails, these adducts induce double-strand breaks (DSBs) that necessitate constant intervention by the Fanconi Anaemia (FA) DNA repair pathway. In the absence of efficient repair, these lesions trigger permanent genomic instability. For the INNERSTANDIN community, it is vital to recognise that this is not merely a localized hepatic issue; acetaldehyde crosses the blood-brain barrier and circulates systemically, causing widespread damage to haematopoietic stem cells, which can lead to bone marrow failure and leukaemogenesis.
Furthermore, the ALDH2 variant interacts destructively with the methionine cycle and cellular methylation signatures. High concentrations of acetaldehyde have been shown to inhibit the activity of DNA methyltransferases (DNMTs) and deplete levels of S-adenosylmethionine (SAMe), the primary universal methyl donor. This disruption precipitates global DNA hypomethylation and site-specific hypermethylation of tumour suppressor genes, creating an epigenetic landscape primed for oncogenesis. In the UK context, evidence from the UK Biobank suggests that even moderate alcohol consumption in ALDH2-deficient individuals significantly elevates the risk of oesophageal squamous cell carcinoma (ESCC) and hypertension, as the inability to clear aldehydes leads to chronic oxidative stress and the activation of pro-inflammatory signalling pathways like NF-κB. This cascade represents a total failure of biological resilience, where a singular genetic substitution transforms a common lifestyle exposure into a profound driver of multi-organ proteotoxicity and accelerated cellular senescence. The INNERSTANDIN perspective demands an acknowledgement that for the ALDH2-deficient cohort, there is no "safe" threshold of exposure; the metabolic machinery is fundamentally unequipped to prevent the resultant DNA vandalism.
What the Mainstream Narrative Omits
While public health discourse in the United Kingdom frequently centres on the macro-physiological consequences of ethanol consumption—such as hepatic steatosis or cardiovascular hyper-reactivity—the mainstream narrative remains dangerously reductive regarding the ALDH2*2 polymorphism. This genetic variant, characterized by a glutamate-to-lysine substitution at position 487 (rs671), is often dismissed as a benign "flushing" reaction. At INNERSTANDIN, our synthesis of the current literature suggests a far more insidious reality: the ALDH2*2 allele represents a profound systemic failure in endogenous and exogenous detoxification, transforming a common beverage into a potent mutagenic assault.
The primary omission in general medical advice is the catastrophic accumulation of acetaldehyde, not merely as a transient metabolite but as a Class 1 carcinogen that actively induces DNA-protein crosslinks (DPCs). Peer-reviewed evidence, most notably from the Medical Research Council (MRC) Laboratory of Molecular Biology, demonstrates that when ALDH2 is compromised, acetaldehyde evades clearance and directly attacks the integrity of haematopoietic stem cells. Research published in *Nature* (Garaycoechea et al., 2018) elucidates that in the absence of functional ALDH2, the cell’s secondary line of defence—the Fanconi Anaemia (FA) DNA repair pathway—is forced into constant, high-intensity activation to rectify double-strand breaks. For individuals carrying the ALDH2 variant, the systemic burden is not merely a "sensitivity"; it is a state of chronic chromosomal instability.
Furthermore, the mainstream narrative ignores the synergy between ALDH2 deficiency and the exhaustion of the NAD+ pool. The oxidation of ethanol and the subsequent (albeit stunted) attempt to oxidise acetaldehyde consume significant quantities of nicotinamide adenine dinucleotide (NAD+). In an ALDH2-deficient landscape, the resulting NADH/NAD+ redox imbalance inhibits sirtuin activity and dysregulates the methionine cycle. This creates a cascade effect where DNA methylation patterns are altered, potentially silencing tumour-suppressor genes. At INNERSTANDIN, we track these epigenetic shifts as critical markers of biological aging.
Moreover, the "glow" reaction is actually a visible manifestation of systemic microvascular inflammation and histaminergic dysregulation. While the NHS often focuses on unit consumption, for the ALDH2-deficient individual, there is no "safe" lower limit. The persistence of N2-ethyl-2'-deoxyguanosine (N2-Et-dG) adducts signifies that even moderate exposure initiates a pro-carcinogenic environment, particularly within the oesophageal and upper aerodigestive epithelia. The failure to categorise ALDH2 deficiency as a definitive contraindication for alcohol consumption represents a significant gap in preventative genomic medicine. To achieve true INNERSTANDIN of this pathology, one must recognise that for the carrier, ethanol is not a toxin to be metabolised, but a direct catalyst for genomic erosion.
The UK Context
Within the geoclinical landscape of the United Kingdom, the prevailing narrative surrounding alcohol consumption often bypasses the nuanced genomic architecture that dictates individual metabolic capacity. At INNERSTANDIN, we recognise that the *ALDH2* polymorphism (specifically the rs671 SNP) represents one of the most significant genetic determinants of systemic toxicity, yet its prevalence in the UK is frequently underestimated due to shifting demographic complexities and the historic "Eurocentric" bias in genomic screening. While the *ALDH2*2 allele is most concentrated in East Asian populations, the UK’s diverse genomic tapestry—enriched by significant South-East Asian and mixed-ancestry cohorts—demands a more rigorous interrogation of aldehyde dehydrogenase deficiency within the NHS clinical framework.
The biological reality is one of profound enzymatic failure. In individuals carrying the *ALDH2*2 variant, a single glutamic acid-to-lysine substitution (Glu504Lys) results in a nearly complete loss of catalytic activity in the mitochondrial ALDH2 enzyme. This creates a metabolic bottleneck where acetaldehyde, a highly reactive and genotoxic intermediate, is not converted into inert acetate. In the UK context, where "binge drinking" culture is statistically prevalent (Office for National Statistics), the inability to clear acetaldehyde leads to immediate cellular crisis. Peer-reviewed data from the UK Biobank has increasingly linked these metabolic deficiencies to heightened risks of oesophageal squamous cell carcinoma and cardiovascular complications, even at "moderate" intake levels.
The mechanism of damage is twofold: direct DNA adduction and epigenetic dysregulation. Acetaldehyde induces the formation of N2-ethylidene-2'-deoxyguanosine (N2-ethylidene-dG) DNA adducts. These lesions, if not resolved by the Fanconi Anemia (FA) DNA repair pathway, precipitate double-strand breaks and chromosomal rearrangements. Furthermore, from the perspective of methylation—a core pillar of INNERSTANDIN research—excess acetaldehyde interferes with the methionine cycle. It inhibits methionine synthase and depletes S-adenosylmethionine (SAMe) levels, thereby compromising DNA methyltransferase (DNMT) activity. This induces a state of global hypomethylation alongside site-specific hypermethylation, fundamentally altering gene expression patterns and accelerating biological ageing. For the UK population, where alcohol-related liver disease (ARLD) is a leading cause of premature mortality, understanding the *ALDH2* status is not merely a matter of "intolerance" but a critical assessment of an individual’s genomic integrity and long-term oncogenic risk. The evidence is unequivocal: for those with the variant, there is no "safe" limit, as the biological machinery required for detoxification is fundamentally structurally compromised.
Protective Measures and Recovery Protocols
The mitigation of acetaldehyde-induced systemic toxicity in individuals carrying the *ALDH2*2* polymorphism requires a multi-layered biochemical strategy that prioritises the acceleration of metabolic clearance and the aggressive sequestration of reactive electrophiles. For the INNERSTANDIN practitioner, the objective is to circumvent the enzymatic bottleneck by bolstering alternative detoxification pathways and fortifying the DNA repair machinery against the inevitable formation of DNA adducts, such as $N^2$-ethyl-2'-deoxyguanosine ($N^2$-Et-dG).
The primary line of biological defence involves the replenishment of the hepatic glutathione (GSH) pool. Acetaldehyde exhibits a high affinity for thiol groups, rapidly depleting intracellular GSH and leaving the cell vulnerable to lipid peroxidation and mitochondrial dysfunction. Clinical research suggests that pre-emptive administration of N-acetyl cysteine (NAC) serves as a critical rate-limiting precursor for GSH synthesis, providing a nucleophilic decoy for acetaldehyde. However, NAC must be integrated with exogenous L-theanine and L-glutamine to maintain the redox homeostasis of the hepatocyte. Furthermore, the flavonoid Dihydromyricetin (DHM), derived from *Hovenia dulcis*, has demonstrated significant efficacy in peer-reviewed trials for its ability to upregulate the expression of both alcohol dehydrogenase (ADH) and residual ALDH activity, effectively reducing the half-life of circulating acetaldehyde in the blood.
From a micronutrient perspective, the recovery protocol must address the inevitable depletion of B-vitamin complexes, particularly Thiamine (B1) and Folate (B9). Thiamine is a mandatory cofactor for the pyruvate dehydrogenase complex; its depletion, exacerbated by the metabolic priority given to ethanol, leads to an accumulation of lactic acid and impaired aerobic respiration. More critically, for those with the *ALDH2* variant, ethanol consumption interferes with the methionine cycle. The resulting inhibition of methionine synthase leads to a reduction in S-adenosylmethionine (SAMe), the universal methyl donor. This compromise in methylation capacity, coupled with acetaldehyde’s direct interference with DNA methyltransferases (DNMTs), necessitates the inclusion of methylcobalamin and 5-MTHF to preserve epigenetic stability and prevent global DNA hypomethylation—a known precursor to ethanol-related carcinogenesis.
Recovery protocols must also facilitate the Nucleotide Excision Repair (NER) and Base Excision Repair (BER) pathways. Magnesium and Zinc act as essential cofactors for the polymerases and nucleases responsible for excising acetaldehyde-derived DNA adducts. Zinc, in particular, is structural to the alcohol dehydrogenase enzyme itself; a deficiency here further impairs the primary metabolic step, creating a deleterious feedback loop of toxicity. Finally, the use of high-dose Vitamin C and Alpha-Lipoic Acid (ALA) is recommended to neutralise the Reactive Oxygen Species (ROS) generated during the induction of the microsomal ethanol-oxidising system (MEOS), specifically the CYP2E1 pathway, which becomes more prominent when the ALDH enzyme is saturated or defective. By synchronising these interventions, the INNERSTANDIN approach transforms a genetic vulnerability into a managed biochemical profile, minimising long-term mutagenic risk.
Summary: Key Takeaways
Alcohol catabolism is governed by a precarious enzymatic relay; however, for those carrying the *ALDH2*2* variant (rs671), this relay collapses at the critical transition from acetaldehyde to acetate. Acetaldehyde is not merely a transient metabolite but a potent, IARC-classified Class 1 carcinogen capable of inducing profound genomic erosion. Peer-reviewed evidence published in *Nature* and *The Lancet* underscores that the incapacity to sequester acetaldehyde results in an influx of DNA-protein cross-links and double-strand breaks, specifically within haematopoietic stem cell populations, precipitating marrow failure and oncogenesis. At INNERSTANDIN, we recognise that the physiological manifestation of the 'flush response' is the overt symptomatic proxy for a covert, systemic mutational burden. In the UK context, where alcohol-related morbidity remains a significant public health burden, the *ALDH2* polymorphism represents a critical gene-environment interaction that renders even 'moderate' consumption hazardous. The cumulative DNA damage—characterised by N2-ethyl-2'-deoxyguanosine adducts—establishes a pro-inflammatory, pro-carcinogenic milieu that extends far beyond hepatic tissue, impacting cardiovascular integrity and neurodegenerative trajectories. This biological reality mandates a shift from viewing alcohol intolerance as a sensitivity toward acknowledging it as a definitive metabolic failure that accelerates cellular senescence and genomic instability.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Alcohol Metabolism and the ALDH2 Variant: The Biological Basis for Intolerance and DNA Damage"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper


