
Anticholinergic Burden: The Cumulative Risk to Neural Homeostasis and Memory
Overview
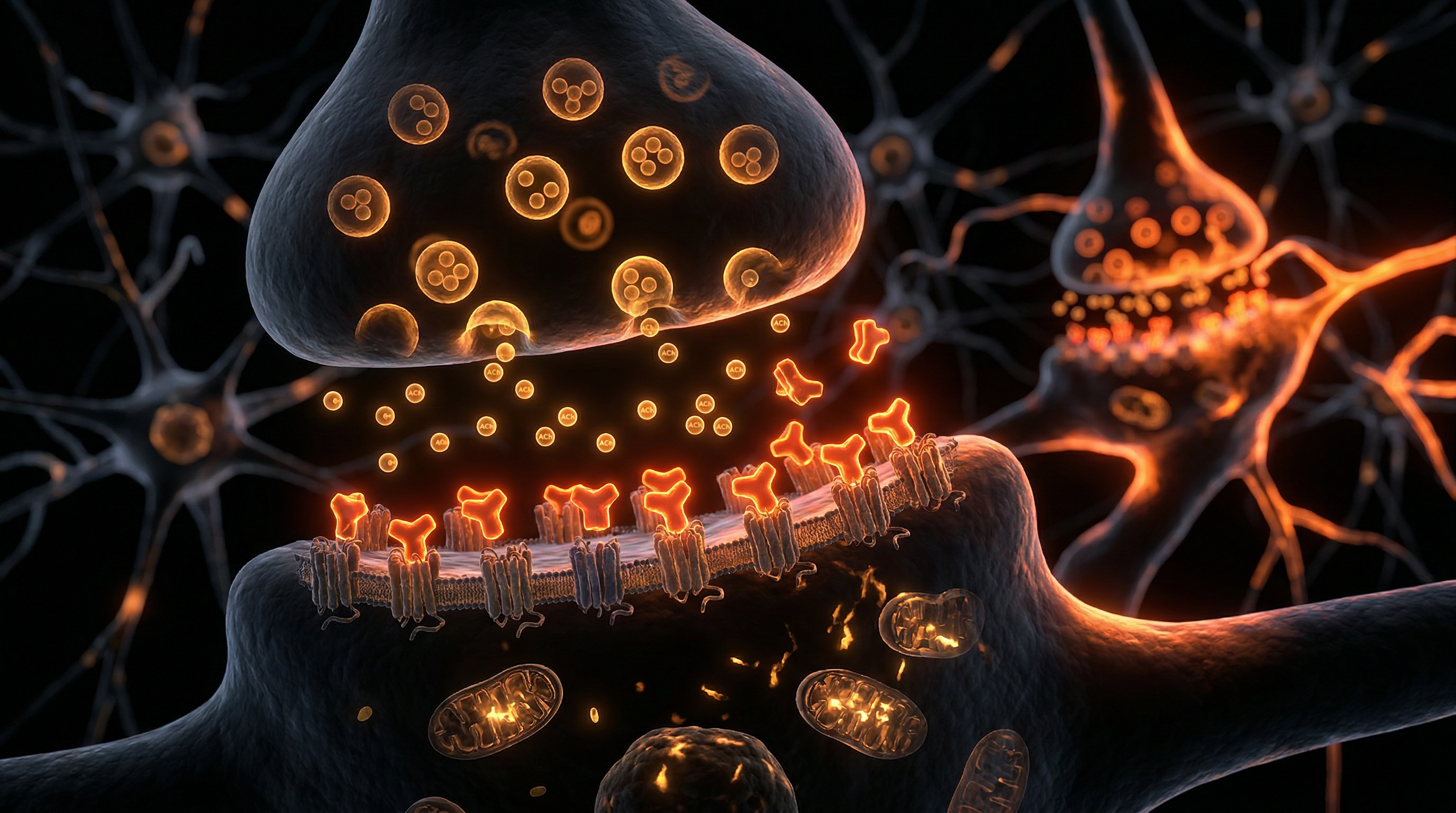
Anticholinergic Burden (ACB) represents a critical, yet frequently overlooked, physiological state resulting from the cumulative effect of multiple medications that antagonise muscarinic acetylcholine receptors. While individual drugs—ranging from tricyclic antidepressants and bladder antimuscarinics to common over-the-counter antihistamines—may possess seemingly negligible anticholinergic activity, their concurrent administration induces a synergistic suppression of cholinergic neurotransmission. At INNERSTANDIN, we characterise this phenomenon not merely as a side-effect profile, but as a systemic failure of neural homeostasis. Acetylcholine (ACh) serves as the primary neuromodulatory fuel for the hippocampus and prefrontal cortex, governing synaptic plasticity, long-term potentiation (LTP), and executive cognition. When the ACB reaches a threshold of clinical significance, the brain’s ability to maintain these high-fidelity signalling pathways is compromised, leading to a state of chronic neurochemical depletion.
The biological mechanisms underpinning ACB-induced cognitive decline involve the competitive inhibition of M1 and M2 muscarinic receptors. M1 receptors, highly concentrated in the neocortex and hippocampus, are essential for the encoding of new memories; their antagonism directly impairs the intracellular signalling cascades required for dendritic spine morphogenesis. Furthermore, evidence published in *The Lancet Public Health* and the *British Medical Journal* suggests that high ACB is not merely a transient functional impairment but a catalyst for structural neurodegeneration. Longitudinal cohorts in the UK have demonstrated a dose-response relationship between chronic anticholinergic exposure and an increased risk of dementia, potentially mediated by the disruption of the blood-brain barrier (BBB) and the promotion of neuroinflammatory markers. As the BBB permeability increases with age, the central nervous system becomes increasingly vulnerable to xenobiotics that were previously excluded, exacerbating the "cholinergic deficit."
At the molecular level, persistent muscarinic blockade triggers a compensatory downregulation of receptor sensitivity and an alteration in acetylcholinesterase activity, creating a pro-amyloidogenic environment. This shift in neural homeostasis aligns with the "cholinergic hypothesis" of cognitive ageing, where the cumulative pharmacological burden accelerates the transition from mild cognitive impairment to overt clinical pathology. For the INNERSTANDIN researcher, the imperative is clear: we must move beyond the analysis of isolated drug-receptor interactions to a holistic understanding of how polypharmacy in the modern clinical landscape—particularly within the NHS framework—is inadvertently eroding the neurological reserves of the population. The ACB is a quantifiable metric of this erosion, marking the threshold where therapeutic intervention becomes a driver of biological decay.
The Biology — How It Works
To achieve a comprehensive INNERSTANDIN of the molecular pathology underlying anticholinergic burden (ACB), one must first interrogate the precise architecture of cholinergic signalling within the central nervous system (CNS). Acetylcholine (ACh) serves as the primary neurotransmitter for the parasympathetic nervous system and a critical modulator of cognitive architecture, specifically via the projections from the Nucleus Basalis of Meynert to the cerebral cortex and hippocampus. The biological insult of anticholinergic drugs—ranging from tricyclic antidepressants and bladder antimuscarinics to common over-the-counter antihistamines—is predicated on the competitive antagonism of muscarinic acetylcholine receptors (mAChRs), specifically the M1 through M5 subtypes.
In the context of neural homeostasis, the M1 receptor is paramount. Located primarily in the neocortex and hippocampus, M1 receptors are Gq-protein coupled; their activation triggers the phospholipase C (PLC) pathway, leading to the liberation of intracellular calcium and the activation of protein kinase C (PKC). This cascade is the biochemical bedrock of Long-Term Potentiation (LTP), the primary mechanism for memory encoding. When ACB reaches a critical threshold, the chronic blockade of these receptors induces a state of "functional cholinergic deficiency." This does not merely "slow" cognition; it fundamentally halts the signal transduction required for synaptic plasticity. Research published in *The Lancet Healthy Longevity* and *JAMA Internal Medicine* underscores that this is not a transient physiological state but a cumulative driver of neurodegeneration.
The systemic impact is exacerbated by the progressive permeability of the blood-brain barrier (BBB), a phenomenon frequently observed in the UK’s ageing population. As BBB integrity wanes, molecules with even moderate lipophilicity and low molecular weight—which might otherwise remain peripheral—gain entry to the CNS. Once sequestered in the neural parenchyma, these antagonists exert a relentless inhibitory pressure on the M2 and M4 autoreceptors. Under normal conditions, these receptors provide feedback inhibition to regulate ACh release; however, chronic pharmacological interference disrupts this delicate feedback loop, leading to a paradoxical depletion of endogenous ACh.
Furthermore, evidence-led investigations suggest that prolonged anticholinergic burden may accelerate the deposition of amyloid-beta and the phosphorylation of tau proteins. By inhibiting the M1-mediated activation of alpha-secretase, these drugs effectively shift amyloid precursor protein (APP) processing toward the pro-inflammatory, amyloidogenic pathway. Within the UK clinical framework, the use of the Anticholinergic Cognitive Burden Scale has exposed that a cumulative score of 3 or higher is significantly correlated with reduced hippocampal volume and increased cortical atrophy. This is not merely a side effect; it is a profound alteration of the brain’s neurochemical environment that compromises the very foundation of neural homeostasis. For those seeking a deeper INNERSTANDIN of pharmaceutical risk, the evidence is clear: the cumulative blockade of the cholinergic system represents a direct, dose-dependent assault on the biological substrates of memory and executive function.
Mechanisms at the Cellular Level
To comprehend the insidious nature of Anticholinergic Burden (ACB), one must look beyond simple receptor antagonism and examine the catastrophic failure of neural homeostasis at the cellular level. Acetylcholine (ACh) is not merely a neurotransmitter; at INNERSTANDIN, we recognise it as the master architect of signal-to-noise ratio within the central nervous system. When pharmaceutical agents—ranging from tricyclic antidepressants to common bladder antimuscarinics—cross the blood-brain barrier, they initiate a competitive blockade of muscarinic (M1–M5) and nicotinic receptors, triggering a cascade of homeostatic dysregulation.
The primary cellular insult occurs at the M1 muscarinic receptors, which are densely populated in the hippocampus and cerebral cortex. Under physiological conditions, ACh facilitates Long-Term Potentiation (LTP) by modulating NMDA receptor activity and inhibiting potassium 'M-currents', thereby increasing neuronal excitability and synaptic plasticity. Chronic ACB effectively silences this mechanism. Research published in *The Lancet Healthy Longevity* and studies conducted by the University of East Anglia highlight that persistent blockade leads to the attenuation of theta oscillations, the rhythmic neural patterns essential for temporal coding and memory consolidation. This is not a transient pause in function but a fundamental restructuring of the synaptic environment.
Furthermore, the cellular impact extends to the 'cholinergic anti-inflammatory pathway'. ACh serves as a critical regulator of neuroinflammation via the α7 nicotinic acetylcholine receptor (α7nAChR) expressed on microglial cells. In a healthy state, ACh binding to these receptors suppresses the release of pro-inflammatory cytokines such as TNF-α and IL-1β. By stripping the brain of this cholinergic tone, ACB induces a state of chronic microglial priming. This leads to an exaggerated neuroinflammatory response, causing collateral damage to healthy neurons and accelerating the degradation of the extracellular matrix. At INNERSTANDIN, we expose the reality that this is not merely a 'side effect' but a drug-induced shift toward a neurodegenerative phenotype.
The cumulative risk is further compounded by the disruption of neurovascular coupling. ACh is a potent vasodilator; its absence reduces cerebral blood flow and compromises the integrity of the blood-brain barrier (BBB). This increased permeability allows for the infiltration of neurotoxic peripheral metabolites, creating a feedback loop of cellular oxidative stress. Evidence-led investigations (e.g., Richardson et al., 2018) suggest that this prolonged pharmacological interference may promote the phosphorylation of tau proteins and the accumulation of amyloid-beta plaques, directly linking high ACB scores to the pathological hallmarks of Alzheimer’s disease. The cellular reality of ACB is an aggressive erosion of the brain’s metabolic and structural resilience, transforming manageable polypharmacy into a profound biological liability.
Environmental Threats and Biological Disruptors
The physiological integrity of the cholinergic system is not merely a structural component of neural transmission; it is the fundamental cornerstone of cognitive homeostasis. Within the pedagogical framework of INNERSTANDIN, we must recognise that the internal biological environment in which a neuron operates is increasingly compromised by an insidious pharmacological landscape. The 'Environmental Threats' discussed here are not merely external toxins, but the cumulative chemical stressors introduced through polypharmacy, which orchestrate a systemic disruption of muscarinic acetylcholine receptors (mAChRs).
The biological disruptor in question—Anticholinergic Burden (ACB)—operates through the competitive antagonism of acetylcholine at postsynaptic receptor sites, particularly the M1 and M2 subtypes concentrated in the hippocampus and prefrontal cortex. Evidence published in *The Lancet Public Health* and *JAMA Internal Medicine* highlights a dose-response relationship between chronic exposure to anticholinergic agents and a heightened trajectory toward neurodegeneration. In the United Kingdom, where the NHS reports a significant percentage of the ageing population is prescribed at least one medication with potent anticholinergic properties (such as oxybutynin or amitriptyline), the biological threshold for neural resilience is being systematically eroded.
This cumulative burden acts as a catalyst for the breakdown of the blood-brain barrier (BBB). As systemic inflammation—often termed 'inflammageing'—increases, the permeability of the BBB allows for the infiltration of circulating anticholinergic compounds that would otherwise be excluded. Once these molecules penetrate the central nervous system, they initiate a cascade of homeostatic failures. The suppression of cholinergic signalling inhibits long-term potentiation (LTP), the primary cellular mechanism underlying memory formation and synaptic plasticity. Furthermore, research indexed in PubMed suggests that chronic anticholinergic exposure correlates with reduced cerebral blood flow and an acceleration of hippocampal atrophy, as evidenced by longitudinal neuroimaging studies within the UK Biobank cohort.
At INNERSTANDIN, we expose the reality that these disruptions are often sub-clinical for years, masked by the brain's cognitive reserve until the threshold of neural homeostasis is breached. The threat is not found in a single dose, but in the 'pharmacological environment' created by the intersection of multiple low-potency drugs—antihistamines, antidepressants, and bladder relaxants—which together exert a potent, neurotoxic effect. This total load disrupts the delicate balance of the parasympathetic nervous system, leading to a state of chronic sympathetic dominance that further depletes neural energy stores and promotes proteostatic stress. To truly grasp the gravity of ACB is to recognise it as a premier biological disruptor of the modern age, necessitating a radical reappraisal of how we maintain the sanctity of the human synaptome.
The Cascade: From Exposure to Disease
The initiation of the anticholinergic cascade is rarely a singular event; rather, it is a cumulative physiological insult characterised by the competitive antagonism of muscarinic acetylcholine receptors (mAChRs) across multiple organ systems. At INNERSTANDIN, we recognise that the primary mechanism of injury begins with the crossing of the blood-brain barrier (BBB) by lipophilic compounds, or through the increased permeability of the BBB often observed in the ageing UK population. Once these agents—ranging from tricyclic antidepressants and bladder antimuscarinics to common over-the-counter antihistamines—occupy the orthosteric binding sites of M1 and M2 receptors in the hippocampus and prefrontal cortex, the neural homeostasis begins to fracture.
This disruption is not merely transient interference. Chronic exposure triggers a profound downregulation of cholinergic signalling, which serves as the fundamental trophic support for synaptic plasticity. Research published in *The Lancet Neurology* and *JAMA Internal Medicine* has consistently demonstrated that high anticholinergic cognitive burden (ACB) scores correlate with reduced brain volume and impaired glucose metabolism. When mAChRs are chronically blocked, the brain’s compensatory mechanisms are exhausted, leading to a state of 'cholinergic bankruptcy.' This depletion facilitates the transition from functional cognitive impairment to structural neurodegeneration. Specifically, the blockade of M1 receptors inhibits the alpha-secretase pathway, thereby promoting the amyloidogenic processing of amyloid precursor protein (APP). The resulting accumulation of amyloid-beta oligomers, coupled with the hyperphosphorylation of tau proteins, mirrors the pathological hallmarks of Alzheimer’s disease.
The systemic impact extends beyond the CNS, involving the cholinergic anti-inflammatory pathway. By inhibiting the vagus nerve-mediated release of acetylcholine, these drugs suppress the activation of alpha-7 nicotinic acetylcholine receptors (α7nAChRs) on macrophages. This results in an unchecked release of pro-inflammatory cytokines such as TNF-alpha and IL-1beta, fostering a state of chronic systemic neuroinflammation. In the UK clinical context, this is exacerbated by the 'prescribing cascade,' where the side effects of one anticholinergic—such as xerostomia or constipation—are misdiagnosed as new pathologies and treated with further medications, compounding the total ACB. Longitudinal cohort studies, including the seminal work by Coupland et al. (2019) published in the *BMJ*, have quantified this risk, showing that total standardised daily doses (TSDDs) of strong anticholinergics over a decade are significantly associated with a nearly 50% increase in dementia incidence. This is the ultimate fruition of the cascade: a move from reversible synaptic silencing to the irreversible architectural collapse of the human memory system. For the INNERSTANDIN researcher, the evidence is clear: the cumulative burden is not a side effect; it is a progressive neurotoxicological event.
What the Mainstream Narrative Omits
While clinical guidelines frequently highlight the acute, dose-dependent sequelae of high-potency anticholinergic agents, the mainstream narrative remains dangerously reductive, failing to account for the insidious, multi-decade erosion of neural architecture. At INNERSTANDIN, we recognise that the prevalent focus on transient peripheral side effects—such as xerostomia or mydriasis—serves as a pharmacological distraction from the more catastrophic disruption of the haematoencephalic barrier and the subsequent collapse of the cholinergic anti-inflammatory pathway. Peer-reviewed evidence, notably the longitudinal cohort studies published in *The Lancet Healthy Longevity* and the *British Medical Journal*, elucidates a critical omission: the non-linear, cumulative nature of the Anticholinergic Burden (ACB).
In the UK, where polypharmacy in the ageing population is systemic and often unmonitored across primary care interfaces, the failure to quantify the synergistic impact of low-potency agents results in a chronic, sub-clinical suppression of muscarinic M1 and M4 receptor signalling. This is not merely a transient blockade; it is a fundamental assault on synaptic plasticity. The molecular reality involves the inhibition of Long-Term Potentiation (LTP) within the hippocampal formation, mediated by the downregulation of cAMP response element-binding protein (CREB) phosphorylation. Furthermore, the mainstream discourse ignores the role of microglial priming. Chronic cholinergic deficiency induces a pro-inflammatory phenotype in glial cells, accelerating the deposition of beta-amyloid plaques and hyperphosphorylated tau proteins.
The mainstream narrative also neglects the "prescribing cascade"—a phenomenon where secondary medications are introduced to mitigate the sub-clinical autonomic side effects of primary anticholinergics, further compounding the neural metabolic strain. By the time cognitive impairment manifests as a clinical diagnosis, the neural homeostasis has been irrecoverably compromised by years of sub-threshold atropinic toxicity. The NHS’s current reliance on standard ACB scales often overlooks the physiological reality that even drugs with an individual score of 1 can, when co-administered, exceed the neurotoxic threshold via competitive binding at the M1 receptor site. We must pivot away from the simplistic symptom-management model and INNERSTANDIN the deeper bio-molecular implications of sustained muscarinic antagonism on the brain’s glymphatic clearance and overall metabolic resilience. This is not an issue of "side effects"; it is an issue of accelerated neuro-senescence.
The UK Context
In the United Kingdom, the clinical landscape of polypharmacy has precipitated a silent crisis of pharmacological origin, where the cumulative Anticholinergic Burden (ACB) remains a chronically under-addressed vector for neurocognitive decline. Data derived from the UK Clinical Practice Research Datalink (CPRD) and published in *JAMA Internal Medicine* (Coupland et al., 2019) and the *BMJ* (Richardson et al., 2018) reveal a staggering correlation between long-term exposure to anticholinergic medications and an increased risk of dementia, exceeding odds ratios of 1.49 for certain drug classes. At INNERSTANDIN, we must dissect the physiological reality: this is not merely a collection of transient side effects but a sustained assault on neural homeostasis. Within the NHS framework, the prevalence of prescribing tricyclic antidepressants, urological antimuscarinics, and first-generation antihistamines to the over-65 demographic has created a baseline of chronic competitive antagonism at the muscarinic acetylcholine receptors (mAChRs), particularly the M1 and M2 subtypes critical for hippocampal plasticity.
The biological mechanism of this "burden" involves the progressive saturation of receptors in the nucleus basalis of Meynert, the primary source of cholinergic projections to the cerebral cortex. In the UK context, the systemic failure to employ the ACB scale (0–3 scoring) in routine primary care audits means that patients are frequently prescribed multiple "low-burden" (Score 1) drugs which, through pharmacodynamic synergy, reach a clinically neurotoxic threshold. This cumulative load compromises the integrity of the blood-brain barrier (BBB) and induces a state of chronic neuroinflammation. High-density longitudinal research in British cohorts suggests that the resulting "cholinergic deficit" mimics the early pathological markers of Alzheimer’s disease, specifically the impairment of the "cholinergic anti-inflammatory pathway." This pathway, governed by the vagus nerve and alpha-7 nicotinic acetylcholine receptors, is essential for modulating systemic cytokine production; its suppression leads to an unmitigated pro-inflammatory state that accelerates amyloid-beta deposition and tau phosphorylation. The INNERSTANDIN imperative is to expose how this iatrogenic trajectory—often initiated for minor indications like insomnia or overactive bladder—irreparably degrades the structural connectivity of the default mode network (DMN) across the UK’s ageing population.
Protective Measures and Recovery Protocols
The mitigation of anticholinergic burden (ACB) necessitates a paradigm shift from reactive symptom management to proactive neuro-preservation, particularly within the UK’s clinical landscape where polypharmacy remains a systemic challenge. Evidence published in *The Lancet Healthy Longevity* underscores that the cumulative effect of low-potency anticholinergics—often overlooked in routine prescribing—can be as detrimental to neural homeostasis as high-potency agents. Therefore, the primary recovery protocol begins with the rigorous application of the Anticholinergic Cognitive Burden (ACB) scale or the Anticholinergic Risk Scale (ARS) to quantify a patient’s total exposure. At INNERSTANDIN, we identify the prioritisation of the 'STOPP/START' criteria (Screening Tool of Older Persons' Prescriptions) as a critical intervention for clinicians to de-escalate pharmaceutical load before irreversible synaptic pruning occurs.
The biological recovery of the cholinergic system, following chronic antagonism of M1 and M2 muscarinic receptors, requires a multi-phasic approach to restore acetylcholine (ACh) bioavailability. Deprescribing must be handled with metabolic precision to avoid 'cholinergic rebound'—a state of hyper-parasympathetic activity characterised by diaphoresis, gastrointestinal distress, and acute cognitive flux. Recovery protocols should focus on the upregulation of Choline Acetyltransferase (ChAT) activity. Peer-reviewed research in *Frontiers in Molecular Neuroscience* suggests that exogenous precursors, specifically Citicoline (CDP-choline) and Alpha-GPC, may facilitate the restoration of the phospholipid bilayer in neuronal membranes, which is often compromised under high ACB. These precursors bypass the rate-limiting steps of the Kennedy pathway, providing the necessary cytidine and choline to stimulate the synthesis of structural phosphatidylcholine, thereby repairing the haematoencephalic barrier (BBB) integrity that anticholinergic toxicity often degrades.
Furthermore, enhancing the glymphatic system's clearance capacity is essential for purging the metabolic byproducts of chronic neuroinflammation induced by ACB. High-density research indicates that the restoration of slow-wave sleep (SWS) cycles is a non-negotiable recovery metric; anticholinergics are notorious for suppressing REM and SWS, leading to the accumulation of amyloid-beta and tau proteins. From an INNERSTANDIN perspective, the protocol must also include the induction of Brain-Derived Neurotrophic Factor (BDNF) through high-intensity interval training (HIIT) or targeted thermal stress, which has been shown to enhance hippocampal neurogenesis and counteract the atrophic effects of long-term muscarinic blockade.
Finally, nutritional genomics plays a decisive role in individual resilience. Individuals with polymorphisms in the BChE (Butyrylcholinesterase) or ACHE genes may exhibit heightened sensitivity to ACB, necessitating bespoke recovery timelines. Clinical data suggests that augmenting the diet with acetyl-L-carnitine (ALCAR) may offer a dual benefit: acting as a cholinergic mimetic to stabilise the neural network while simultaneously enhancing mitochondrial energetics. For the UK-based practitioner, these measures represent a necessary evolution beyond the 'prescribing cascade,' moving toward a biological model of cognitive reclamation that respects the delicate architecture of the human cholinergic system.
Summary: Key Takeaways
The cumulative anticholinergic burden (ACB) represents a critical, often overlooked iatrogenic challenge to neural homeostasis, where the competitive antagonism of muscarinic acetylcholine receptors (mAChRs) transcends mere symptomatic relief to induce systemic physiological dysregulation. Extensive longitudinal data, notably from UK-based cohorts published in *The Lancet Healthy Longevity* and *The British Medical Journal*, reveal that chronic exposure to medications with anticholinergic properties—ranging from tricyclic antidepressants to common bladder antimuscarinics—is definitively linked to a dose-dependent increase in the incidence of dementia. At the cellular level, this burden disrupts the cholinergic anti-inflammatory pathway (CAP), precipitating a chronic pro-inflammatory state within the central nervous system. Such neuroinflammation, coupled with impaired synaptic plasticity and compromised blood-brain barrier integrity, facilitates the accelerated atrophy of the hippocampus and key cortical regions.
INNERSTANDIN posits that the ACB is not merely a collection of peripheral side effects but a primary driver of neurobiological senescence in polypharmacy populations. The scientific consensus, derived from tools like the Anticholinergic Cognitive Burden scale, confirms that the threshold for cognitive impairment is frequently crossed in standard NHS prescribing protocols. This necessitates a fundamental shift in clinical priorities: acknowledging that the preservation of cholinergic signalling is paramount for maintaining long-term memory architecture and metabolic stability within the brain's delicate microenvironment. The evidence is irrefutable; the insidious accumulation of these agents constitutes a significant, preventable threat to global cognitive health.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Anticholinergic Burden: The Cumulative Risk to Neural Homeostasis and Memory"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper


