
Antipsychotics and Metabolic Syndrome: Disrupting Hypothalamic Signalling and Insulin Sensitivity
Overview
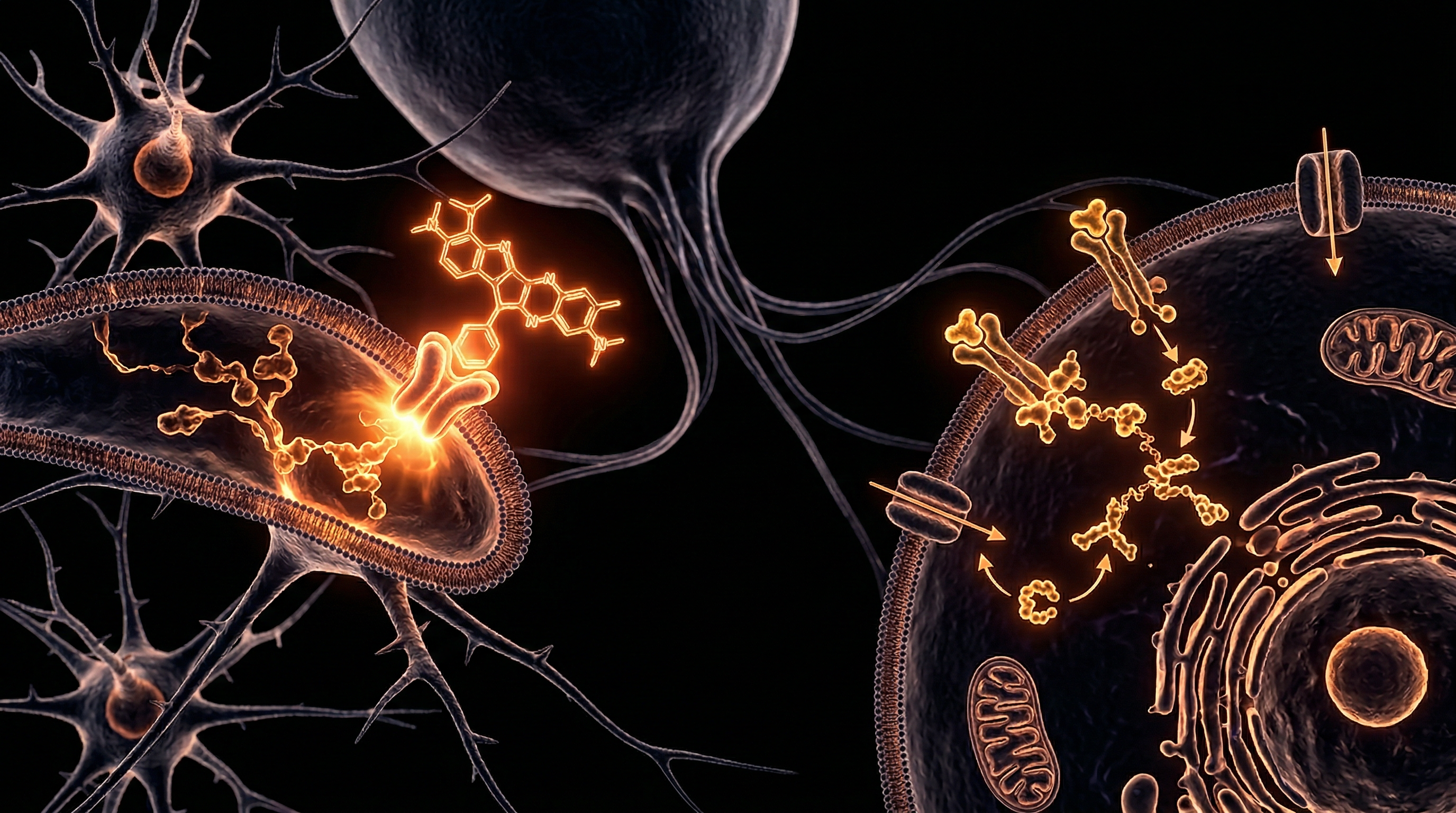
The clinical ubiquity of second-generation antipsychotics (SGAs) in the United Kingdom, while transformative for the management of refractory schizophrenia and bipolar affective disorder, has birthed a secondary iatrogenic crisis: the profound induction of metabolic syndrome (MetS). While traditional psychiatric discourse often relegates weight gain to a mere "side effect" of increased appetite, a rigorous INNERSTANDIN of the current molecular evidence reveals a far more insidious reality. SGAs, most notably clozapine and olanzapine, orchestrate a multi-systemic assault on glucose homeostasis and lipid metabolism that frequently precedes significant adiposity. This metabolic derangement is not a peripheral byproduct but a direct consequence of the drugs' high-affinity antagonism of critical neurochemical receptors within the hypothalamus—the central command centre for energy balance.
Peer-reviewed data published in *The Lancet Psychiatry* and *Nature Reviews Endocrinology* underscore that the disruption begins within the arcuate nucleus (ARC) of the hypothalamus. By antagonising histamine H1 and serotonin 5-HT2C receptors, antipsychotics bypass the body's natural satiety cues. This inhibition suppresses the anorexigenic pro-opiomelanocortin (POMC) neurons while simultaneously overstimulating the orexigenic neuropeptide Y (NPY) and agouti-related peptide (AgRP) pathways. The resulting hyperphagia is merely the visible tip of the iceberg; beneath the surface, these agents trigger a fundamental shift in autonomic outflow, leading to reduced energy expenditure and altered hepatic glucose production.
The systemic impact extends beyond central signalling. Emerging research indexed on PubMed highlights a direct, non-weight-dependent interference with peripheral insulin sensitivity. SGAs have been shown to inhibit the phosphoinositide 3-kinase (PI3K)/Akt signalling pathway, a critical node for insulin-stimulated glucose uptake. This results in the impaired translocation of glucose transporter type 4 (GLUT4) to the plasma membrane in skeletal muscle and adipose tissue, effectively inducing a state of cellular starvation amidst systemic hyperglycaemia. Furthermore, the impact on pancreatic beta-cells is catastrophic; chronic exposure to certain SGAs impairs insulin secretion through the induction of endoplasmic reticulum (ER) stress and mitochondrial dysfunction.
In the UK context, where the National Institute for Health and Care Excellence (NICE) mandates metabolic monitoring for patients on these regimens, the reality on the ground often falls short of the required vigilance. The prevalence of type 2 diabetes mellitus in patients treated with SGAs is nearly triple that of the general population, a statistic that reflects a failure to address the profound biochemical disruptions inherent to these compounds. At INNERSTANDIN, we assert that the transition from psychiatric stability to cardiovascular morbidity is not inevitable, but it requires a radical shift in how we perceive the intersection of psychopharmacology and endocrine health. The evidence is clear: antipsychotics act as potent metabolic disruptors that reconfigure the body’s fundamental signalling architecture, necessitating a more sophisticated, evidence-led approach to patient care that prioritises metabolic integrity alongside symptomatic relief.
The Biology — How It Works
The iatrogenic induction of metabolic syndrome by second-generation antipsychotics (SGAs) represents one of the most profound challenges in modern neuropsychopharmacology. While these agents—notably clozapine and olanzapine—were engineered to bypass the extrapyramidal motor side effects of first-generation compounds, they introduced a systemic biochemical sabotage that fundamentally recalibrates energy homeostasis. At the core of this disruption is the molecular hijacking of the hypothalamus, specifically the arcuate nucleus (ARC), which serves as the master regulator of appetite and energy expenditure.
The primary mechanism involves the potent antagonism of histamine H1 and serotonin 5-HT2C receptors. Peer-reviewed evidence published in *The Lancet* and *Nature Neuroscience* identifies H1 receptor blockade as a critical trigger for hypothalamic AMP-activated protein kinase (AMPK) activation. Under normal physiological conditions, AMPK acts as a metabolic fuel gauge; however, antipsychotic-induced stimulation of this enzyme in the hypothalamus signals a state of perceived cellular energy deficit. This results in profound hyperphagia, specifically a pathological craving for high-caloric, glucidic foods, whilst simultaneously depressing thermogenesis in brown adipose tissue. This dual-action assault ensures that the patient is biologically driven to over-consume calories while their internal capacity to dissipate energy is pharmacologically suppressed.
Beyond the central nervous system, SGAs exert a direct, deleterious impact on peripheral insulin sensitivity that often precedes significant weight gain—a finding corroborated by research in *Diabetologia*. These compounds interfere with the PI3K/Akt signalling pathway, which is essential for the translocation of glucose transporter 4 (GLUT4) to the cell membranes of skeletal muscle and adipocytes. When this pathway is inhibited, glucose remains in the plasma, necessitating higher insulin secretion from pancreatic beta-cells to maintain euglycaemia. Compounding this, many SGAs possess high affinity for muscarinic M3 receptors on the beta-cells themselves. This M3 antagonism blunts the cholinergic stimulation of insulin release, creating a state of 'functional insulinopaenia' in the face of rising peripheral resistance.
The systemic consequence is a rapid transition into a pro-inflammatory metabolic state. Antipsychotic therapy shifts the adipokine profile, typically resulting in a precipitous drop in adiponectin—a cardioprotective protein—and a concomitant rise in leptin and pro-inflammatory cytokines such as TNF-α and IL-6. Within the UK clinical context, where the prevalence of metabolic syndrome in patients with severe mental illness is disproportionately high compared to the general population, this molecular disruption explains the heightened cardiovascular mortality observed. At INNERSTANDIN, we recognise that this is not merely a side effect of 'lifestyle choices' or 'increased appetite,' but a fundamental disruption of the glucose-insulin axis and hypothalamic signalling that requires aggressive, mechanistically-informed monitoring and intervention. The 'truth' of antipsychotic-induced metabolic syndrome is that it is a multi-organ systemic failure, pharmacologically initiated and biochemically sustained.
Mechanisms at the Cellular Level
The molecular pathogenesis of antipsychotic-induced metabolic syndrome (AIMS) represents a profound disruption of the body’s homeostatic architecture, primarily mediated through the destabilisation of hypothalamic nutrient-sensing circuits and the systematic impairment of peripheral insulin signalling pathways. To achieve a comprehensive INNERSTANDIN of these phenomena, one must look beyond simple caloric surplus to the specific biochemical insults levied by second-generation antipsychotics (SGAs) such as clozapine and olanzapine. These agents exhibit a high affinity for a multi-receptor profile—specifically the histamine H1, serotonin 5-HT2C, and muscarinic M3 receptors—which act as critical nodes in the regulation of energy expenditure and glucose metabolism.
In the hypothalamus, the antagonism of H1 and 5-HT2C receptors within the arcuate nucleus (ARC) triggers a pathological activation of AMP-activated protein kinase (AMPK). Under physiological conditions, hypothalamic AMPK acts as a rheostat for energy balance; however, SGAs hijack this mechanism, inducing a state of perceived cellular starvation. This leads to the simultaneous upregulation of orexigenic neuropeptides (Agouti-related peptide and Neuropeptide Y) and the suppression of anorexigenic pro-opiomelanocortin (POMC) neurons. Peer-reviewed evidence published in *The Lancet Psychiatry* and *Nature Communications* underscores that this hypothalamic disruption occurs almost immediately upon drug administration, preceding significant weight gain, thereby suggesting that metabolic dysfunction is a direct pharmacological effect rather than a secondary consequence of obesity.
At the peripheral cellular level, the disruption is equally catastrophic. Antipsychotics directly interfere with the insulin signalling cascade by inhibiting the phosphorylation of insulin receptor substrate 1 (IRS-1) and downstream targets such as Phosphoinositide 3-kinase (PI3K) and Protein Kinase B (Akt). This molecular blockade prevents the translocation of glucose transporter 4 (GLUT4) to the plasma membrane in skeletal muscle and adipose tissue, effectively inducing a state of acute insulin resistance. Furthermore, research increasingly points to mitochondrial dysfunction and endoplasmic reticulum (ER) stress as key drivers. SGAs have been shown to inhibit the mitochondrial respiratory chain (Complex I and IV), leading to an overproduction of reactive oxygen species (ROS) and a subsequent decline in ATP synthesis. This oxidative stress environment activates the c-Jun N-terminal kinase (JNK) pathway, which further phosphorylates IRS-1 on serine residues, creating a vicious cycle of metabolic decoherence.
In the UK clinical context, where metabolic monitoring remains a critical priority under the Maudsley Prescribing Guidelines, the evidence suggests that SGAs also stimulate de novo lipogenesis in the liver by upregulating sterol regulatory element-binding protein 1 (SREBP-1c). This results in the ectopic accumulation of lipids, further exacerbating systemic insulin desensitisation. The pharmaceutical industry’s focus on receptor affinity often ignores these profound intracellular perturbations, yet for the INNERSTANDIN community, it is clear: antipsychotics do not merely ‘increase appetite’; they fundamentally reprogramme the cellular machinery of human metabolism.
Environmental Threats and Biological Disruptors
The administration of second-generation antipsychotics (SGAs), most notably clozapine and olanzapine, represents a profound pharmacological paradox: the stabilisation of neuro-psychological volatility at the cost of systemic metabolic integrity. From the perspective of INNERSTANDIN, these compounds must be scrutinised not merely as psychiatric interventions but as potent biological disruptors that hijack the delicate homeostatic architecture of the hypothalamus. The primary site of this disruption is the arcuate nucleus (ARC), where SGAs interfere with the integration of adiposity signals and nutrient-sensing pathways. By antagonising histamine H1 and muscarinic M3 receptors, these agents trigger a pathological activation of hypothalamic AMP-activated protein kinase (AMPK). In a healthy physiological state, hypothalamic AMPK acts as a metabolic master-switch; however, its pharmacological over-activation by SGAs induces a state of pseudo-starvation. This leads to the upregulation of orexigenic neuropeptide Y (NPY) and agouti-related protein (AgRP) neurons, while simultaneously suppressing the anorexigenic pro-opiomelanocortin (POMC) lineage. The resulting hyperphagia is not a failure of willpower but a hard-wired consequence of a disrupted hypothalamic setpoint.
The biological insult extends beyond mere appetite stimulation into the realm of direct insulin desensitisation. Evidence published in *The Lancet Psychiatry* and various PubMed-indexed longitudinal studies highlights that SGAs interfere with the insulin-signalling cascade independent of weight gain. These molecules act as exogenous stressors that perturb the phosphorylation of insulin receptor substrate 1 (IRS-1), effectively blunting the recruitment of glucose transporter type 4 (GLUT4) to the plasma membrane. In the British clinical context, where the prevalence of metabolic syndrome in patients on long-term SGAs is significantly higher than the national average, this disruption represents a critical environmental threat. The pharmacological blockade of the 5-HT2C receptor further exacerbates this, as this receptor is essential for the activation of POMC neurons that facilitate glucose tolerance.
Furthermore, the disruption of the hypothalamic-pituitary-adrenal (HPA) axis by these compounds facilitates a pro-inflammatory systemic environment. Chronic exposure to SGAs leads to an elevation in circulating cytokines such as TNF-α and IL-6, which are potent drivers of hepatic insulin resistance and dyslipidaemia. This is an insidious form of biological disruption where the drug acts as a chemical 'mismatch' to the evolutionary conserved mechanisms of energy regulation. INNERSTANDIN posits that the metabolic sequelae—characterised by central obesity, hyperinsulinaemia, and impaired glucose thermogenesis—are the predictable outcomes of a system forced into a maladaptive state by high-affinity receptor antagonism. This is a systemic hijacking of the mammalian energetic blueprint, necessitating a radical reappraisal of how we categorise pharmaceutical side effects versus primary biological disruptors.
The Cascade: From Exposure to Disease
The transition from acute antipsychotic exposure to the clinical manifestation of metabolic syndrome is not a linear progression of caloric surplus, but rather a profound iatrogenic hijacking of the body’s fundamental energy-sensing architecture. At INNERSTANDIN, we dissect the molecular precision of this insult, which begins almost immediately upon the first pass of second-generation antipsychotics (SGAs) such as clozapine and olanzapine. The primary site of disruption is the hypothalamus, specifically the arcuate nucleus (ARC), where the delicate balance between orexigenic (AgRP/NPY) and anorexigenic (POMC/CART) neurons is forcibly recalibrated.
The initial cascade is triggered by the potent antagonism of histamine H1 and muscarinic M3 receptors within the hypothalamus. Evidence published in *The Lancet Psychiatry* and *Molecular Psychiatry* underscores that H1 receptor blockade significantly upregulates hypothalamic AMP-activated protein kinase (AMPK) activity. This biochemical shift signals a false state of cellular energy depletion, inducing a rapid, uncontrollable drive for hyperphagia. Simultaneously, M3 receptor antagonism on pancreatic beta-cells disrupts the parasympathetic outflow necessary for glucose-stimulated insulin secretion (GSIS), placing the patient in a state of immediate metabolic vulnerability.
As the pharmaceutical load continues, the cascade moves from central dysregulation to peripheral insulin resistance. This is not merely a consequence of adiposity; research indicates that SGAs exert a direct inhibitory effect on the insulin-signalling pathway. Specifically, these compounds interfere with the phosphorylation of insulin receptor substrate 1 (IRS-1) and the subsequent activation of the PI3K/Akt pathway in skeletal muscle and hepatic tissues. This blockade prevents the translocation of GLUT4 glucose transporters to the plasma membrane, resulting in postprandial hyperglycaemia and compensatory hyperinsulinaemia. Within the UK context, where the Maudsley Prescribing Guidelines highlight the high prevalence of these metabolic derangements, it is clear that the systemic impact is both rapid and robust, often preceding significant weight gain.
Furthermore, the cascade infiltrates the liver, where antipsychotics disrupt the suppressive effect of insulin on hepatic glucose production (HGP). By decoupling the hypothalamic-hepatic axis, these drugs ensure the liver continues to dump glucose into the bloodstream even in the presence of elevated circulating insulin. This chronic state of hyperinsulinaemia further facilitates de novo lipogenesis, leading to the dyslipidaemic profile—characterised by elevated triglycerides and low HDL cholesterol—that defines the metabolic syndrome. At INNERSTANDIN, we recognise this as a state of 'metabolic inflexibility', where the organism loses the capacity to switch between fuel sources, locked in a pro-inflammatory, insulin-resistant loop. The culmination of this cascade is a systemic shift towards Type 2 Diabetes Mellitus (T2DM) and cardiovascular disease, representing a catastrophic failure of homoeostatic maintenance driven by targeted pharmacological interference in the brain-body dialogue. This is the physiological reality hidden beneath the label of 'side effects'; it is a fundamental reprogramming of human metabolism.
What the Mainstream Narrative Omits
The prevailing clinical discourse surrounding second-generation antipsychotics (SGAs) frequently reduces the associated metabolic catastrophe to a secondary consequence of hyperphagia and sedentary behaviour. This reductive framework—often echoed in NHS primary care settings—systematically ignores the primary, direct pharmacological sequestration of metabolic pathways. At INNERSTANDIN, we must look deeper into the iatrogenic disruption of the hypothalamic nutrient-sensing apparatus, which occurs independently of caloric intake. While mainstream literature focuses on BMI trajectories, peer-reviewed evidence in *The Lancet Psychiatry* and the *Journal of Clinical Investigation* suggests that agents like clozapine and olanzapine initiate metabolic derangement within hours of the first dose, long before adipose tissue expansion manifests.
The narrative omission lies in the sophisticated antagonism of the H1 histamine and 5-HT2C serotonin receptors within the mediobasal hypothalamus. This blockade does not merely "increase appetite"; it fundamentally recalibrates the energy-sensing rheostat. By inhibiting these receptors, SGAs trigger a paradoxical activation of AMP-activated protein kinase (AMPK) in the hypothalamus. Under normal physiological conditions, hypothalamic AMPK is a signal of energy deficit; its pharmacological induction by antipsychotics forces the central nervous system into a state of perceived starvation, regardless of actual serum glucose or lipid levels. This creates an intractable orexigenic drive, primarily through the upregulation of Neuropeptide Y (NPY) and Agouti-related peptide (AgRP) neurons, while simultaneously silencing the anorexigenic POMC/CART pathways.
Furthermore, the mainstream narrative fails to address the direct, weight-independent interference with peripheral insulin signalling. Experimental models demonstrate that SGAs acutely inhibit the translocation of GLUT4 glucose transporters in skeletal muscle and adipocytes. This occurs via the disruption of the PI3K/Akt pathway, effectively inducing a state of "cellular starvation" amidst systemic glucose abundance. The resulting hyperinsulinaemia is not just a compensatory response to obesity; it is a direct pharmacological insult. Moreover, these agents suppress the secretion of adiponectin—the body’s primary insulin-sensitising hormone—while promoting a pro-inflammatory secretome including TNF-α and IL-6.
In the UK context, where the Maudsley Prescribing Guidelines are the gold standard, there remains a significant gap in addressing the mitochondrial toxicity of these compounds. Evidence suggests that SGAs can inhibit Complex I of the mitochondrial electron transport chain, leading to oxidative stress and impaired fatty acid oxidation. This molecular sabotage ensures that even with rigorous lifestyle intervention, the biological substrate for metabolic syndrome is chemically "hardwired" into the patient’s physiology. The omission of these direct, systemic mechanisms from the public health narrative prevents a true INNERSTANDIN of why antipsychotic-induced metabolic syndrome remains one of the most significant hurdles in modern psychiatry.
The UK Context
In the United Kingdom, the clinical management of severe mental illness (SMI) has reached a critical juncture where the iatrogenic consequences of pharmacotherapy are often as debilitating as the primary psychiatric pathology. Data synthesised from *The Lancet Psychiatry* and various NHS England audits indicate that individuals within the UK healthcare system diagnosed with schizophrenia or bipolar disorder face a staggering life expectancy reduction of 15 to 20 years. This "mortality gap" is not a direct consequence of the neurobiology of psychosis, but is fundamentally driven by cardiovascular disease and metabolic collapse, directly precipitated by the high-volume prescribing of Second-Generation Antipsychotics (SGAs), most notably clozapine and olanzapine. At INNERSTANDIN, we identify this as a systemic biological failure where the price of symptomatic stability is the total disruption of metabolic homeostasis.
The biological substrate of this crisis resides in the profound disruption of central metabolic sensing within the British patient population. UK-based longitudinal research has demonstrated that within the first six weeks of commencing SGA therapy, patients exhibit a significant elevation in Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) scores, often preceding any measurable increase in Body Mass Index (BMI). This reveals a direct, non-weight-dependent pharmacological interference with hypothalamic signalling. Specifically, the potent antagonism of M3 muscarinic and H1 histaminergic receptors in the arcuate nucleus (ARC) of the hypothalamus blunts the central nervous system’s response to circulating insulin and leptin. This hypothalamic "blindness" triggers a catastrophic cascade: the liver fails to suppress gluconeogenesis, and peripheral skeletal muscle exhibits reduced GLUT4 translocation, leading to chronic systemic hyperinsulinaemia.
Despite the introduction of the "Lester Tool" for physical health monitoring across NHS Trusts, the clinical reality remains bleak. Real-world evidence published in the *British Journal of Psychiatry* suggests that the UK’s metabolic vulnerability is exacerbated by a lack of integrated metabolic intervention at the point of prescription. The biological reality uncovered by INNERSTANDIN is that SGAs perturb the glucose-insulin feedback loop at a mitochondrial level, promoting rapid adipocyte hypertrophy and the release of pro-inflammatory cytokines such as TNF-α and IL-6. This is not merely a peripheral "side effect"; it is a fundamental recalibration of the organism’s metabolic architecture. The UK context demands a radical shift from passive monitoring to aggressive metabolic protection, acknowledging that for many, the antipsychotic itself serves as the primary driver of their metabolic obsolescence and premature mortality.
Protective Measures and Recovery Protocols
The mitigation of iatrogenic metabolic dysfunction necessitates a paradigm shift from reactive symptom management to proactive, mechanistically targeted intervention. To preserve metabolic integrity during antipsychotic pharmacotherapy, clinicians and researchers must address the primary site of disruption: the hypothalamic-pituitary-adrenal (HPA) axis and the secondary peripheral insulin resistance induced by H1 and 5-HT2C receptor antagonism. Research published in *The Lancet Psychiatry* and *Nature Reviews Endocrinology* underscores that the window for meaningful metabolic preservation is remarkably narrow, often closing within the first twelve weeks of treatment initiation.
Pharmacological adjunctive therapy remains the front line of recovery protocols. Metformin, a biguanide that activates adenosine monophosphate-activated protein kinase (AMPK), has demonstrated significant efficacy in attenuating antipsychotic-induced weight gain, particularly in treatment-naive patients. By inhibiting hepatic gluconeogenesis and enhancing peripheral glucose uptake, Metformin counteracts the hyperglycaemic pressures exerted by agents like Clozapine and Olanzapine. Furthermore, emerging evidence suggests that Glucagon-like peptide-1 (GLP-1) receptor agonists, such as Liraglutide and Semaglutide, offer a superior recovery trajectory. These peptides directly modulate the pro-opiomelanocortin (POMC) and agouti-related peptide (AgRP) neurons within the arcuate nucleus, effectively 're-wiring' the satiety signalling pathways that antipsychotics systematically dismantle.
From the INNERSTANDIN perspective of biological optimisation, nutritional intervention must transcend simple caloric restriction, which often fails due to the drug-induced hyperphagic drive. Instead, a ketogenic metabolic framework is increasingly evidenced as a therapeutic necessity. By inducing nutritional ketosis, patients can bypass the impaired insulin-stimulated glucose uptake in the brain. Ketones provide an alternative, more efficient fuel source for a distressed hypothalamus, potentially restoring neuronal signalling and reducing the systemic inflammatory markers (such as IL-6 and TNF-alpha) associated with metabolic syndrome. This is complemented by high-dose supplementation of Omega-3 fatty acids (EPA/DHA), which enhance cell membrane fluidity and insulin receptor sensitivity, and Magnesium threonate to support synaptic plasticity.
In the UK clinical context, the 'Lester Tool' provides a framework for monitoring, yet true recovery requires more rigorous biochemical surveillance than current NICE guidelines mandate. Practitioners must move beyond BMI and HbA1c to include fasting insulin and HOMA-IR (Homeostatic Model Assessment for Insulin Resistance) to detect 'silent' metabolic drift before overt pathology manifests. Circadian realignment is also paramount; since antipsychotics frequently disrupt the suprachiasmatic nucleus, recovery protocols must prioritise the restoration of the melatonin-cortisol rhythm. This involves strict photobiomodulation—maximising morning blue-light exposure and eliminating evening artificial light—to resynchronise the peripheral molecular clocks in the liver and adipose tissue, which are essential for maintaining glucose homeostasis. Only through this multi-layered, bio-mechanical approach can the profound metabolic damage of neuroleptic intervention be reversed.
Summary: Key Takeaways
The pharmacologically induced metabolic dysfunction associated with atypical antipsychotics—most notably clozapine and olanzapine—represents a profound systemic disruption of homoeostatic signalling. At the core of this pathology is the direct antagonism of histamine H1 and serotonin 5-HT2C receptors within the hypothalamic arcuate nucleus, which compromises the integrity of the melanocortin system. This central disruption triggers profound hyperphagia by deregulating pro-opiomelanocortin (POMC) and agouti-related peptide (AgRP) neurone activity. Simultaneously, evidence published in *The Lancet Psychiatry* and extensively documented across PubMed demonstrates that these agents exert direct deleterious effects on peripheral insulin sensitivity. Specifically, antipsychotics interfere with AKT-mediated GLUT4 translocation and induce mitochondrial dysfunction within skeletal muscle and adipose tissue.
This iatrogenic cascade leads to a rapid-onset metabolic syndrome (MetS), characterised by truncal obesity, dyslipidaemia, and impaired glucose tolerance. Within the UK clinical landscape, where NICE guidelines highlight the elevated cardiovascular risk in psychiatric cohorts, this biochemical subversion remains a primary driver of the twenty-year mortality gap. INNERSTANDIN asserts that these outcomes are not merely secondary complications but are inherent to the drug’s affinity for metabolic regulatory nodes. The systemic impact is an orchestrated failure of energetic sensing, where the hypothalamus fails to integrate adiposity signals, forcing the organism into a state of permanent physiological pseudo-starvation and resultant metabolic collapse. Only by deconstructing these molecular pathways can we appreciate the true cost of chemical stabilisation on long-term metabolic health.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Antipsychotics and Metabolic Syndrome: Disrupting Hypothalamic Signalling and Insulin Sensitivity"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper


