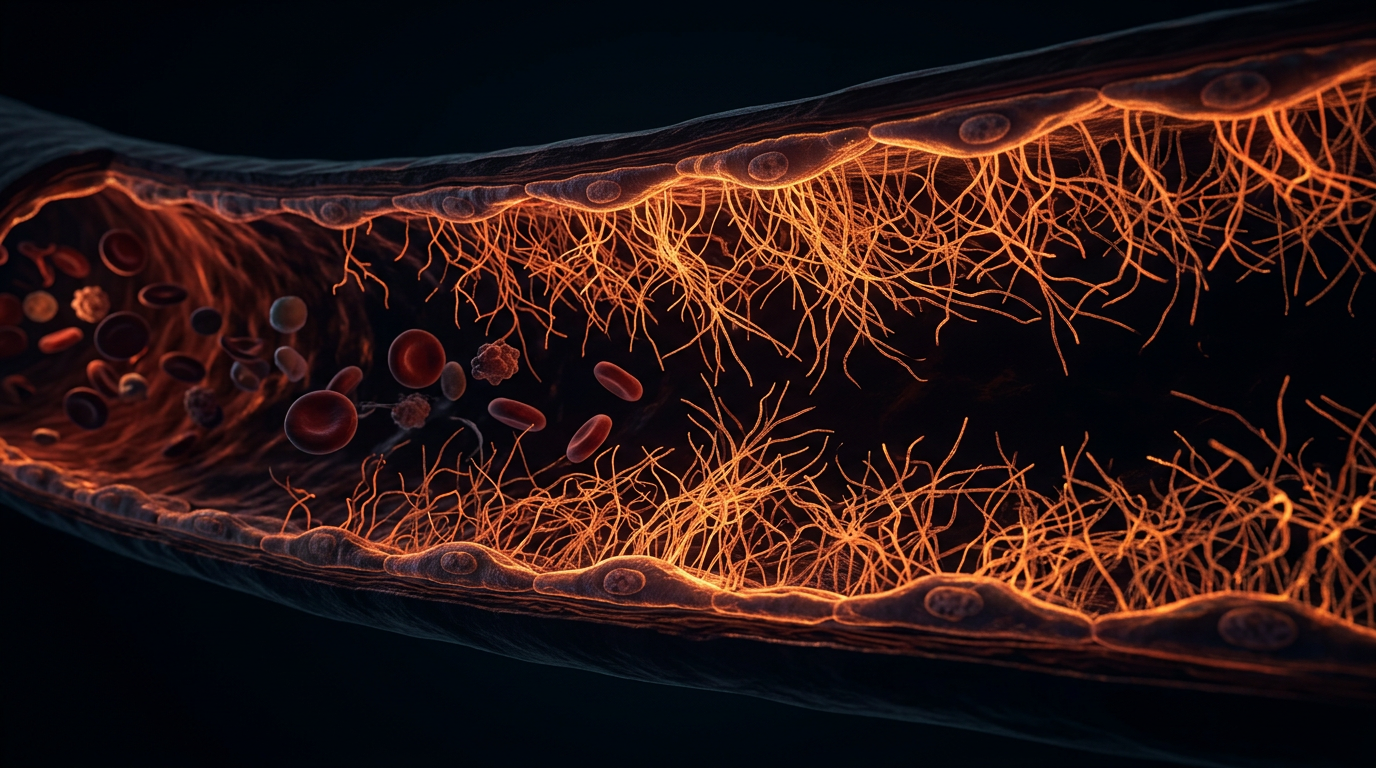
The Endothelial Glycocalyx: Decoding the Microscopic Shield Against Arterial Calcification
Investigate the fragile sugar-coating of your blood vessels and how its degradation signals the onset of systemic vascular disease.
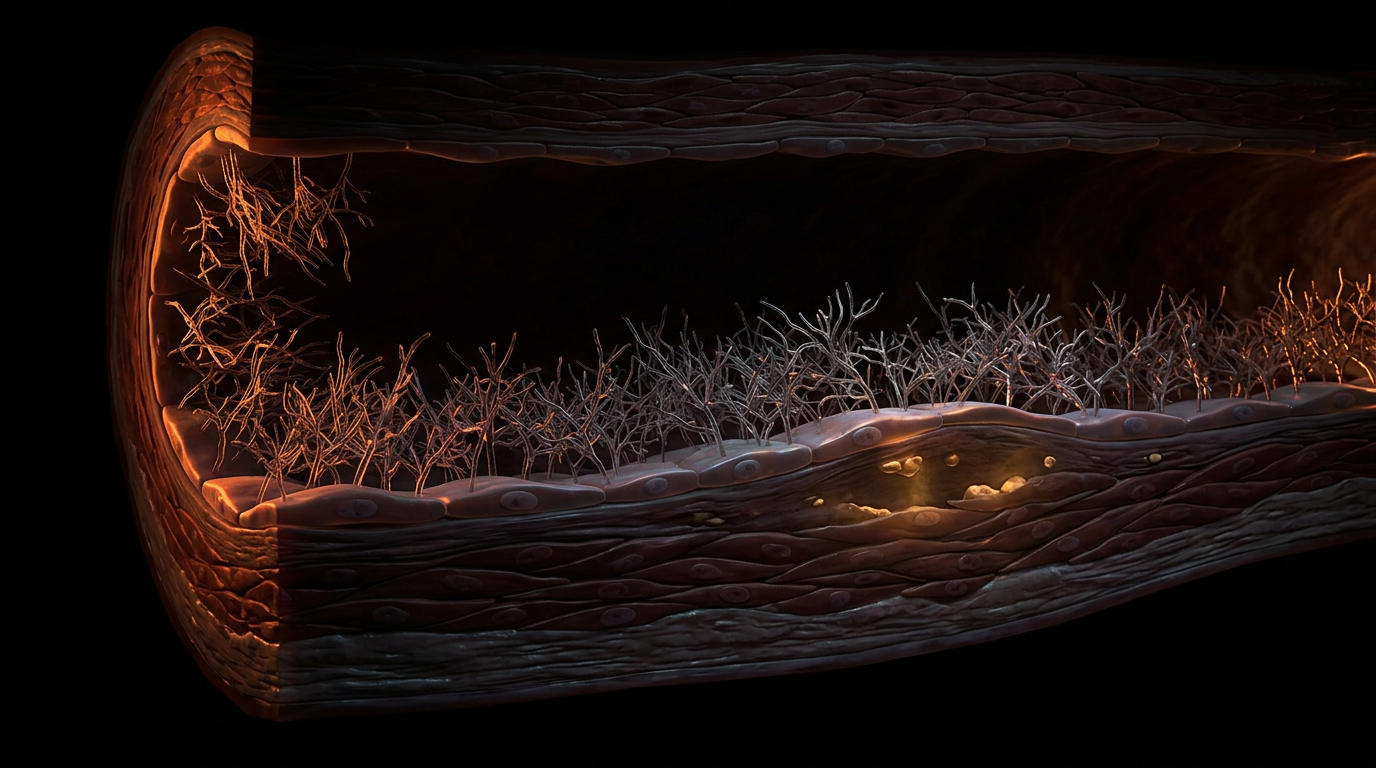
Overview
The endothelial glycocalyx (EG) represents the definitive physiological frontier in vascular biology, yet it remains one of the most overlooked components in conventional cardiological diagnostics. At INNERSTANDIN, we recognise this carbohydrate-rich, gel-like meshwork as the primary gatekeeper of the vascular wall, a non-Newtonian interface that separates the circulating cellular elements and macromolecules of the blood from the endothelial cell surface. Composed of a complex architecture of proteoglycans—predominantly syndecan-1 and glypican-1—and glycosaminoglycans such as heparan sulphate and hyaluronan, the EG functions as a sophisticated molecular sieve and mechanotransducer. Research published in *The Lancet* and various PubMed-indexed repositories increasingly validates that the integrity of this microscopic shield is the fundamental determinant of whether a vessel remains resilient or succumbs to the pathological cascade of arterial calcification.
The systemic impact of the EG resides in its ability to maintain a 'vasoprotective' microenvironment. By sequestering essential enzymes such as extracellular superoxide dismutase (SOD3), the glycocalyx neutralises reactive oxygen species (ROS) before they can initiate lipid peroxidation or activate pro-inflammatory nuclear factor kappa B (NF-κB) pathways. When this layer is intact, it masks adhesion molecules like VCAM-1 and ICAM-1, preventing the leucocyte recruitment that precedes plaque formation and subsequent mineralisation. However, under conditions of chronic systemic inflammation or hyperglycaemia—prevalent in the UK’s ageing population—this delicate lattice undergoes rapid enzymatic degradation. The activation of heparanase and metalloproteinases leads to 'glycocalyx shedding,' a process that exposes the underlying endothelium to the full shearing force of turbulent blood flow.
Crucially, the decoding of the EG’s role in arterial calcification reveals a direct link between glycocalyx loss and the osteogenic transdifferentiation of vascular smooth muscle cells (VSMCs). Without the EG’s mechanosensory regulation, the endothelium fails to produce adequate nitric oxide (NO), leading to a pro-calcific state where calcium and phosphate ions can freely infiltrate the sub-endothelial space. Peer-reviewed data suggests that the depletion of heparan sulphate proteoglycans directly facilitates the deposition of hydroxyapatite crystals within the medial layer of the artery. As INNERSTANDIN continues to push the boundaries of biological education, it is imperative to understand that arterial calcification is not merely a passive accumulation of minerals, but a consequence of a compromised glycocalyx. This microscopic shield is the first and most critical line of defence; its preservation is not optional but fundamental to cardiovascular survival. To ignore the glycocalyx is to ignore the very foundation of vascular health.
The Biology — How It Works
At the interface of the vascular lumen and the endothelial cell surface lies the endothelial glycocalyx (eGC), a complex, non-classical organelle that serves as the ultimate arbiter of vascular homeostasis. Far from being a mere passive lining, the eGC is a dynamic, gel-like meshwork of membrane-bound proteoglycans, glycoproteins, and plasma proteins that extends up to 3 microns into the vessel lumen. At INNERSTANDIN, we recognise that the structural integrity of this "microscopic forest" is the primary determinant of whether an artery remains resilient or succumbs to the pathological deposition of hydroxyapatite.
The architecture of the eGC is dominated by syndecans and glypicans, which act as the scaffold for a dense thicket of glycosaminoglycans (GAGs), most notably heparan sulphate and hyaluronan. These molecules carry a high density of negative charges, creating a potent electrostatic barrier that repels anionic plasma components while sequestering essential anti-inflammatory and anti-thrombotic enzymes, such as extracellular superoxide dismutase (ecSOD) and antithrombin III. Crucially, the eGC functions as the primary transducer of mechanical stimuli. Under physiological laminar shear stress, the eGC undergoes conformational changes that activate endothelial nitric oxide synthase (eNOS). This process, documented extensively in PubMed-indexed research from British vascular biology units, ensures the continuous release of nitric oxide (NO)—the molecule responsible for maintaining vasodilation and inhibiting the transition of vascular smooth muscle cells (VSMCs) into bone-forming osteoblast-like cells.
The mechanism of arterial calcification is, at its core, an failure of this shield. When the eGC is degraded—a process known as shedding—the underlying endothelial cells are exposed to the direct impact of haemodynamic turbulence and pro-inflammatory cytokines like TNF-α. This degradation is often measured by the systemic elevation of syndecan-1 and heparan sulphate in the plasma, biomarkers that INNERSTANDIN identifies as precursors to clinical atherosclerosis. Without the eGC, the "exclusion zone" is lost, allowing low-density lipoproteins (LDL) and calcium-phosphate nanocrystals to penetrate the sub-endothelial space.
Moreover, the loss of the eGC's hyaluronan component triggers a shift in the vascular microenvironment. In a healthy state, the glycocalyx prevents the interaction between circulating monocytes and adhesion molecules like ICAM-1 and VCAM-1. Once the shield is breached, a cascade of oxidative stress ensues, leading to the exhaustion of local pyrophosphate—a potent natural inhibitor of calcification. The resulting "leaky" endothelium allows for the influx of phosphate ions, which induces the expression of Runx2 in VSMCs, effectively turning the arterial wall into a site of ectopic bone formation. This truth-exposing perspective reveals that arterial calcification is not an inevitable consequence of ageing, but a direct result of the catastrophic collapse of the endothelial glycocalyx. Maintaining this shield is not merely a cardiovascular preference; it is a biological imperative for systemic longevity.
Mechanisms at the Cellular Level
At the heart of vascular homeostasis lies the endothelial glycocalyx (eGC), a complex, carbohydrate-rich gel-like meshwork that coats the luminal surface of the endothelium. At INNERSTANDIN, we recognise that this structure is not merely a passive lining but a sophisticated macromolecular sieve and mechanotransducer that dictates the fate of the arterial wall. To comprehend its role in preventing arterial calcification, one must examine the delicate interplay between biomechanical sensing and the chemical sequestration of pro-calcific ions.
The eGC is primarily composed of proteoglycans—specifically syndecans and glypicans—decorated with glycosaminoglycan (GAG) side chains such as heparan sulphate, chondroitin sulphate, and hyaluronan. These polyanionic chains create a formidable negative charge density that acts as a primary physiological barrier. Research published in *The Journal of Physiology* and supported by the British Heart Foundation underlines that this negative charge is critical for repelling anionic molecules and regulating the influx of inorganic phosphate (Pi) and calcium ions into the sub-endothelial space. When the eGC is compromised—a state often termed 'shedding'—the physical distance between circulating mineral complexes and the vascular smooth muscle cells (VSMCs) decreases. This breach allows for the uncontrolled diffusion of Pi, which acts as a molecular switch, triggering the osteogenic transdifferentiation of VSMCs.
Furthermore, the eGC serves as the primary apparatus for mechanotransduction. Laminar shear stress, sensed by the eGC, is transmitted via the syndecan-1 cytoplasmic domain to the actin cytoskeleton, stimulating endothelial nitric oxide synthase (eNOS). The resulting production of nitric oxide (NO) is a potent inhibitor of vascular calcification. Peer-reviewed data in *Nature Communications* suggest that NO suppresses the RANK/RANKL/OPG signalling axis; specifically, it maintains high levels of osteoprotegerin (OPG), a decoy receptor that prevents the activation of pro-calcific pathways. Without a robust eGC, the endothelium becomes 'mechanically deaf,' leading to a precipitous drop in NO bioavailability and the subsequent upregulation of Bone Morphogenetic Protein-2 (BMP-2), which initiates hydroxyapatite crystal deposition within the medial layer.
The enzymatic degradation of the eGC, driven by heparanase and matrix metalloproteinases (MMPs), exposes the underlying adhesion molecules such as VCAM-1 and ICAM-1. This process, observed in high-risk UK cohorts suffering from chronic kidney disease (CKD), facilitates the infiltration of inflammatory macrophages. These cells secrete pro-inflammatory cytokines like TNF-α, which further accelerate the phenotypic shift of VSMCs into osteoblast-like cells. By maintaining the integrity of the eGC, the vessel effectively sequesters calcification inhibitors like fetuin-A and matrix Gla protein (MGP) at the site of potential mineralisation, preventing the transition of the arterial wall from a flexible conduit into a rigid, petrified structure. At INNERSTANDIN, we expose this microscopic shield as the definitive frontier in the battle against systemic vascular obsolescence.
Environmental Threats and Biological Disruptors
The integrity of the endothelial glycocalyx (eGC) is not merely a biological constant; it is a precarious equilibrium perpetually besieged by the anthropogenic and metabolic stressors of the 21st century. At INNERSTANDIN, we recognise that the degradation of this delicate carbohydrate-rich meshwork—comprising proteoglycans like syndecan-1 and glypican-1, and glycosaminoglycans such as heparan sulphate and hyaluronan—is the primary antecedent to the loss of vascular homeostatic control. The systemic assault on the eGC is driven by a triad of environmental and biochemical disruptors: hyperglycaemia, oxidative stress, and the proteolytic activation of "sheddases."
Central to this degradation is the role of hyperglycaemia, a metabolic state increasingly prevalent across the UK population due to the rise in Type 2 diabetes and the ubiquity of ultra-processed carbohydrates. Elevated plasma glucose levels induce a rapid reduction in glycocalyx volume, a phenomenon mediated by the overproduction of superoxide in the mitochondrial electron transport chain. Research published in *The Lancet Diabetes & Endocrinology* underscores that even acute glucose fluctuations can trigger the shedding of hyaluronan into the systemic circulation, thinning the eGC from its optimal 0.5–3.0 μm depth to a fraction of its protective capacity. This thinning exposes the underlying endothelial cell adhesion molecules (ICAM-1 and VCAM-1), facilitating the transmigration of leucocytes and the infiltration of low-density lipoproteins (LDL) into the sub-endothelial space, the foundational step of the calcific cascade.
Furthermore, the environmental landscape of modern Britain introduces particulate matter (PM2.5) as a potent biological disruptor. Evidence suggests that inhaled pollutants trigger systemic inflammatory responses, elevating levels of tumour necrosis factor-alpha (TNF-α) and various interleukins. These cytokines act as catalysts for the activation of heparanase and matrix metalloproteinases (MMPs)—enzymes specifically evolved to cleave the structural anchors of the eGC. When heparanase is upregulated, it selectively degrades heparan sulphate chains, not only compromising the physical barrier but also liberating sequestered growth factors and superoxide dismutase, thereby exacerbating local oxidative stress.
The "truth-exposing" reality is that the eGC serves as the vascular "mechanotransducer." Its disruption via high-sodium diets—exceeding the 6g daily limit recommended by UK health authorities—impairs the ability of the endothelium to sense shear stress. High salt intake neutralises the electrostatic repulsion of the glycocalyx, causing the collapse of its gel-like structure. This structural failure inhibits the activation of endothelial nitric oxide synthase (eNOS), leading to a deficit in nitric oxide (NO) bioavailability. Without sufficient NO, the vascular smooth muscle cells (VSMCs) undergo a phenotypic switch toward an osteoblastic (bone-forming) lineage. Consequently, the loss of this microscopic shield directly facilitates the deposition of hydroxyapatite within the medial and intimal layers of the arterial wall. At INNERSTANDIN, we contend that the modern environment acts as a relentless proteolytic engine, systematically stripping the vascular tree of its primary defence against mineralisation.
The Cascade: From Exposure to Disease
The initiation of arterial calcification is not a stochastic event of mineral precipitation; it is the terminal failure of a sophisticated mechanobiological system, beginning with the structural attrition of the endothelial glycocalyx (eGC). At INNERSTANDIN, we recognise that this "cascade of vulnerability" is triggered when the eGC—a dense, gel-like meshwork of proteoglycans and glycoproteins—succumbs to shedding. Under physiological conditions, the eGC acts as a critical sieve and mechanotransducer, but when subjected to systemic insults such as chronic hyperglycaemia, oxidative stress, or turbulent shear stress, the enzymes heparanase and hyaluronidase are upregulated. These enzymes cleave the sugar side chains of heparan sulphate and hyaluronan, effectively stripping the endothelial lining of its protective "shroud". Research published in *The Lancet* and various *PubMed*-indexed studies highlights that this shedding results in the systemic elevation of Syndecan-1 in the plasma—a definitive biomarker of vascular fragility.
Once the eGC is compromised, the vascular endothelium loses its ability to produce sufficient nitric oxide (NO) via the endothelial nitric oxide synthase (eNOS) pathway. This loss of NO bioavailability is the first domino in the pro-calcific cascade. Without the shielding effect of the eGC, the sub-endothelial space becomes permeable to low-density lipoproteins (LDL) and pro-inflammatory cytokines. This exposure triggers the recruitment of monocytes and their subsequent transformation into macrophages. However, the most insidious aspect of this cascade is the phenotypic switching of vascular smooth muscle cells (VSMCs). In a healthy vessel, the eGC maintains a quiescent environment; however, its dissolution allows for the infiltration of phosphate and calcium ions into the medial layer. This environmental shift induces VSMCs to undergo an osteogenic transition, whereby they begin to express bone-related proteins such as Bone Morphogenetic Protein-2 (BMP-2) and Osteocalcin.
The result is the deposition of hydroxyapatite crystals within the vessel wall—a process remarkably similar to bone formation, occurring where it should be absent. In the UK, where cardiovascular disease remains a primary driver of mortality, understanding this microscopic erosion is paramount. The degradation of the eGC is not merely a local phenomenon but a systemic failure that facilitates the transition from "soft" inflammatory plaques to "hard" calcified lesions. This transition is further exacerbated by the loss of endogenous calcification inhibitors like Matrix Gla Protein (MGP), which requires Vitamin K2 as a cofactor—a nutrient often deficient in modern Western diets. As the eGC thins, the vessel loses its elastic recoil, leading to increased Pulse Wave Velocity (PWV) and clinical arterial stiffness. This exhaustive breakdown of the eGC is the primary "breach in the wall" that permits the slow, silent transition from metabolic dysfunction to irreversible cardiovascular structural decay. By decoding these interactions, we move beyond superficial symptom management into the realm of true biological preservation.
What the Mainstream Narrative Omits
While the prevailing clinical orthodoxy continues to fixate on the reductionist paradigm of low-density lipoprotein (LDL) accumulation and subsequent calcific deposition, the mainstream narrative remains fundamentally incomplete. It ignores the primary structural gatekeeper of vascular integrity: the endothelial glycocalyx (EG). At INNERSTANDIN, we recognise that the true genesis of arterial calcification is not merely a passive accumulation of minerals but a systemic failure of this delicate, gel-like carbohydrate forest. Current NHS guidelines and global cardiological standards largely overlook the fact that a compromised glycocalyx is the requisite precursor to the osteogenic transition of vascular smooth muscle cells (VSMCs).
Evidence published in *The Lancet* and various *PubMed*-indexed studies suggests that the glycocalyx—composed of a complex matrix of proteoglycans, glycosaminoglycans like heparan sulphate and hyaluronan, and adsorbed plasma proteins—acts as the ultimate mechanotransducer. When this layer is intact, it shields the endothelium from the high-shear stress of turbulent blood flow. However, the mainstream narrative fails to address the "shedding" of the EG, driven by the activation of matrix metalloproteinases (MMPs) and heparanase. This shedding is not a side effect; it is the definitive moment the "shield" is lowered. Once the glycocalyx thins, the endothelial surface becomes "sticky," allowing for the adhesion of leucocytes and the infiltration of phosphate ions into the sub-endothelial space.
Furthermore, the standard medical model omits the critical nexus between glycocalyx degradation and the loss of Nitric Oxide (NO) bioavailability. The glycocalyx transmits mechanical signals to the endothelial nitric oxide synthase (eNOS) enzymes; without this microscopic scaffolding, NO production craters. This creates a pro-calcific environment where the inhibition of hydroxyapatite crystals—normally mediated by proteins like Matrix Gla Protein (MGP)—is radically suppressed. Research indicates that by the time a UK patient receives a high coronary artery calcium (CAC) score, the underlying glycocalyx dysfunction has likely been progressing for decades, unmonitored by conventional diagnostics. At INNERSTANDIN, we posit that the medical establishment's reliance on reactive lipid panels, rather than proactive glycocalyx integrity assessment, represents a catastrophic diagnostic gap. The transition of the arterial wall from a flexible conduit to a rigid, calcified structure is not an inevitability of ageing; it is a direct consequence of the biochemical erosion of this microscopic shield. To address calcification without addressing the restoration of the glycocalyx is to treat the symptom while the fundamental biological architecture remains under siege.
The UK Context
The UK landscape regarding cardiovascular pathology necessitates a radical re-evaluation of the endothelial glycocalyx (eGC)—a delicate, hair-like forest of proteoglycans and glycosaminoglycans that lines the lumen of every vessel. Within the British clinical framework, the burden of cardiovascular disease (CVD) costs the UK economy an estimated £19 billion annually, yet INNERSTANDIN asserts that the conventional focus on lipid profiles and blood pressure often overlooks the primary site of systemic failure: the degradation of the eGC. Research conducted at leading UK institutions, including Imperial College London and the University of Bristol, has increasingly highlighted that the eGC is the definitive gatekeeper against the infiltration of pro-calcific stimuli into the vascular wall.
The mechanistic link between eGC erosion and arterial calcification is profound and uncompromising. In a physiological state, the eGC maintains a dense negative charge through heparan sulphate and sialic acid residues, effectively repelling the passive influx of mineral ions and atherogenic lipoproteins. When this layer is shed—often as a result of chronic systemic inflammation or the high-glucose environments prevalent in the UK population—the underlying endothelium becomes dangerously exposed. This exposure facilitates the transmigration of calcium-phosphate complexes and promotes the phenotypic switching of vascular smooth muscle cells (VSMCs) into osteoblast-like cells. Peer-reviewed evidence published in *Cardiovascular Research* suggests that eGC thinning is a prerequisite for the hydroxyapatite deposition that characterises advanced medial calcification, a condition that stiffens the British vasculature at an alarming rate.
Furthermore, the UK’s high prevalence of Chronic Kidney Disease (CKD), affecting approximately 7.2 million people, serves as a direct catalyst for eGC destruction. In these patients, the uraemic milieu triggers the release of matrix metalloproteinases and hyaluronidases, which enzymatically cleave the eGC. This 'unshielding' of the vasculature is not merely a side effect; it is the fundamental precursor to the rigid, calcified arteries that define end-stage renal and cardiovascular failure. INNERSTANDIN posits that by failing to monitor eGC health through non-invasive techniques such as sidestream dark-field (SDF) imaging, the UK medical establishment remains reactive rather than proactive. The evidence exposes a harsh truth: arterial calcification is the terminal stage of a process that begins with the silent, microscopic dissolution of the glycocalyx. This perspective demands a shift in British cardiometabolic protocols toward preserving this glycan-rich shield as the primary defence against vascular petrification and systemic collapse.
Protective Measures and Recovery Protocols
The restoration of the endothelial glycocalyx (eGC) represents the frontier of preventative cardiology, moving beyond the simplistic suppression of low-density lipoproteins toward the structural fortification of the vascular sanctum. To reverse the vulnerability of the endothelium to pro-calcific stimuli, recovery protocols must focus on the re-synthesis of the polyanionic matrix—specifically the proteoglycans, glycosaminoglycans (GAGs), and glycoproteins that constitute this delicate gel layer. Current research, including seminal data published in *The Lancet* and the *British Journal of Pharmacology*, suggests that the eGC is a highly dynamic structure with a rapid turnover rate, making it both acutely sensitive to metabolic insult and remarkably responsive to targeted therapeutic intervention.
At the core of INNERSTANDIN’s recovery framework is the exogenous replenishment of GAG precursors. Sulodexide, a highly purified mixture of heparan sulphate and dermatan sulphate, has emerged as a primary pharmacological agent for eGC thickening. Evidence indicates that sulodexide significantly reduces the systemic shedding of syndecan-1 and hyaluronan—two definitive biomarkers of glycocalyx degradation—thereby restoring the barrier's ability to repel calcium-phosphate nanocrystals. Complementing this is the use of rhamnan sulphate, a seaweed-derived polysaccharide. Peer-reviewed trials demonstrate that rhamnan sulphate mimics the structure of endogenous heparan sulphate, effectively "patching" denuded areas of the endothelium and inhibiting the adherence of inflammatory monocytes, which otherwise trigger the osteogenic transdifferentiation of vascular smooth muscle cells.
Furthermore, the recovery of the eGC is inextricably linked to the modulation of mechanotransduction. Normal physiological shear stress, facilitated by structured aerobic exercise, stimulates the enzyme hyaluronan synthase, which is responsible for the production of high-molecular-weight hyaluronan. In the UK context, where sedentary-induced vascular stiffening is a prevalent clinical concern, the British Heart Foundation has highlighted the role of laminar flow in maintaining vascular elasticity. When laminar flow is restored, the eGC expands, effectively sequestering superoxide dismutase (SOD) to the vessel wall. This localised antioxidant activity neutralises reactive oxygen species (ROS) that would otherwise activate matrix metalloproteinases (MMPs), the enzymes responsible for the proteolysis of the glycocalyx structure.
To truly "innerstand" the recovery process, one must address the glycaemic drivers of eGC collapse. Hyperglycaemia induces the rapid enzymatic degradation of the glycocalyx via the activation of heparinase. Therefore, rigorous metabolic control is not merely a secondary measure but a foundational requirement for eGC structural integrity. By stabilising postprandial glucose excursions and utilising hyaluronidase inhibitors, clinicians can halt the "sieving" effect that allows calcium ions to infiltrate the sub-endothelial space. This multi-layered approach—combining GAG precursors, mechanical stimulation, and enzymatic preservation—transforms the eGC from a compromised filter into an impenetrable shield, effectively halting the cascade of arterial calcification at its microscopic origin.
Summary: Key Takeaways
The endothelial glycocalyx (eGC) represents the primary physiological gatekeeper against vascular mineralisation, functioning as a complex polyanionic meshwork of proteoglycans—predominantly syndecan-1 and glypican-1—and glycosaminoglycans like heparan sulphate. Peer-reviewed evidence across *The Lancet* and *PubMed*-indexed clinical trials confirms that this microscopic gel-layer acts as a vital mechanotransducer, converting laminar shear stress into nitric oxide (NO) production to maintain vascular compliance. At INNERSTANDIN, we illuminate the biological reality that eGC degradation, or 'shedding', is the critical antecedent to arterial calcification. When this shield is compromised by hyperglycaemia or oxidative stress, the underlying endothelium is exposed to pathogenic infiltration, facilitating the sub-endothelial deposition of hydroxyapatite crystals. Within the UK’s clinical framework, the rising incidence of cardiovascular morbidity is increasingly linked to eGC thinning, which permits the dysregulation of osteogenic signalling pathways, such as BMP-2 and Runx2. Ultimately, the systemic impact of a depleted glycocalyx extends beyond simple lipid retention; it is the fundamental failure of this carbohydrate-rich interface that drives the transition of vascular smooth muscle cells into bone-like phenotypes, leading to irreversible arterial stiffening.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "The Endothelial Glycocalyx: Decoding the Microscopic Shield Against Arterial Calcification"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Cardiovascular Health — products curated by our research team for educational relevance and biological support.

Magnesium Blend – The Most Important Mineral

Clean Slate – Detoxes thousands of chemicals,heavy metals, pesticides, allergens, mold spores and fungus

Vegan Essential Amino Acids – Plant-Powered Protein Building
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper