
The Nephron's Precision: How the Kidneys Regulate Blood pH and Electrolyte Balance
The kidneys are far more than simple filters; they are the master chemists of the human body. Gain insight into how the nephron maintains the delicate balance of fluids and electrolytes necessary for life.
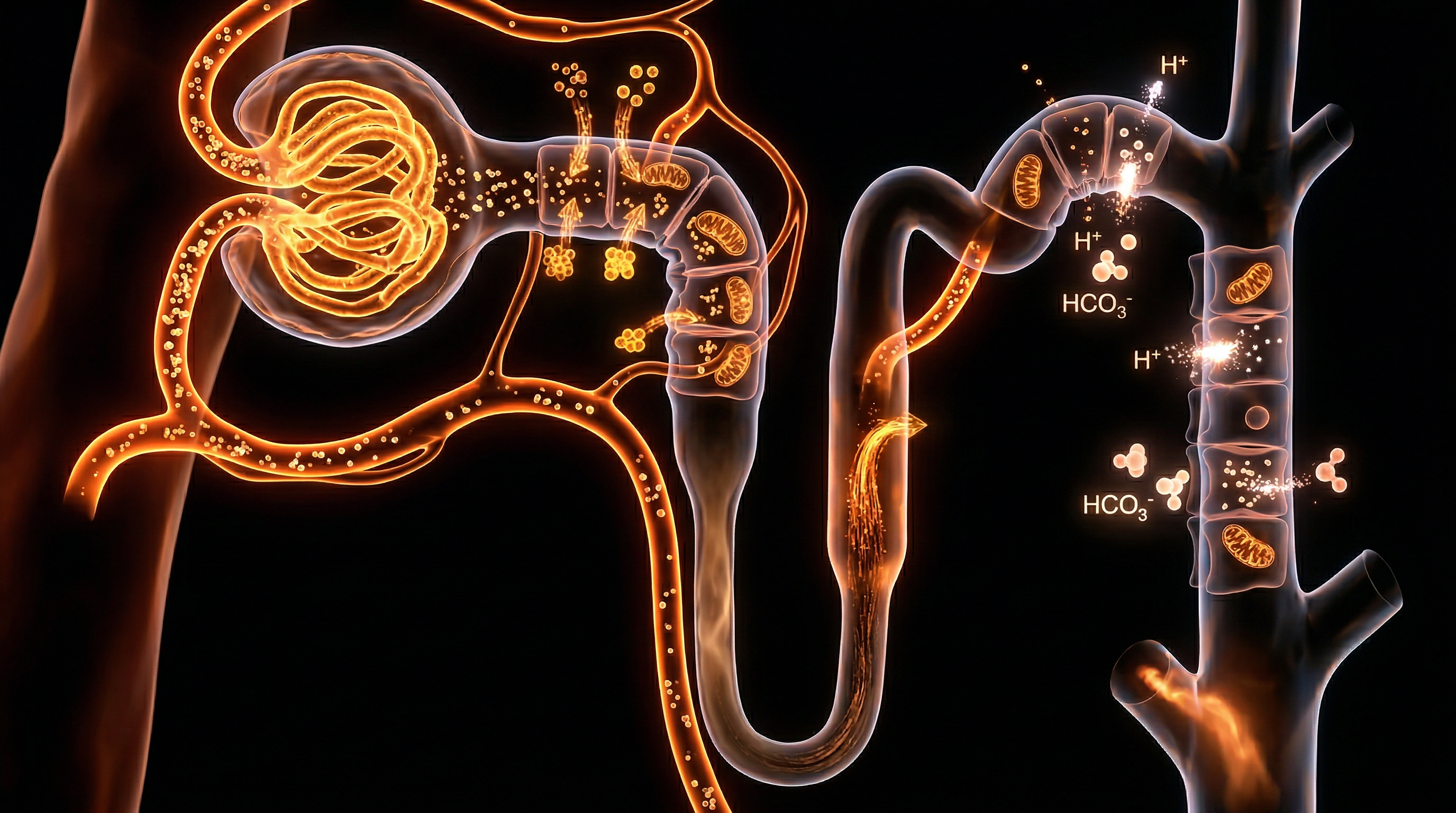
Overview
The renal system represents the ultimate arbiter of internal milieu stability, a feat of biological engineering managed by the nephron with exquisite metabolic fidelity. While often colloquially dismissed as a mere filtration apparatus for nitrogenous waste, the nephron is, in truth, a sophisticated sensory and effector architecture that dictates the physicochemical boundaries of human life. To achieve a profound INNERSTANDIN of renal physiology, one must acknowledge that every cellular process—from the propagation of neuronal action potentials to the kinetics of enzymatic catalysis—is fundamentally contingent upon the kidney’s ability to maintain arterial blood pH within the narrowest of physiological windows (7.35–7.45) and electrolytes within precise millimolar concentrations.
The architectural complexity of the nephron—comprising the glomerulus, the proximal convoluted tubule (PCT), the loop of Henle, the distal convoluted tubule (DCT), and the collecting duct—facilitates a hierarchical, segmental approach to homeostasis. According to seminal research published in *Nature Reviews Nephrology*, the PCT is responsible for the bulk reabsorption of approximately 80–90% of filtered bicarbonate ($HCO_3^-$), a process intrinsically linked to the secondary active transport of hydrogen ions ($H^+$) via the sodium-hydrogen exchanger 3 (NHE3). This is not a passive recovery; it is an energy-intensive reclamation project that prevents the catastrophic acidification of the extracellular fluid.
Furthermore, the nephron’s precision extends to the meticulous calibration of potassium ($K^+$), sodium ($Na^+$), and calcium ($Ca^{2+}$), where the distal segments exercise "fine-tuning" capabilities under hormonal duress. The renin-angiotensin-aldosterone system (RAAS), a cornerstone of clinical nephrology within the UK’s NHS frameworks, modulates the epithelial sodium channels (ENaC) and the $Na^+/K^+$-ATPase pump to regulate blood pressure and serum tonicity. As highlighted in *The Lancet*, even minor deviations in this electrolytic orchestration—such as hyperkalaemia—can precipitate lethal cardiac arrhythmias, underscoring the kidney's role as the primary safeguard against systemic metabolic collapse.
The "truth-exposing" reality of renal function lies in its dual-purpose mechanism: the simultaneous titration of volatile and non-volatile acidity alongside the calibration of ionic strength. Through the synthesis of ammonium ($NH_4^+$) and the generation of *de novo* bicarbonate, the kidneys compensate for the daily "acid load" produced by protein metabolism. This overview establishes that the nephron is not merely a biological filter, but a dynamic, self-regulating processor that ensures the systemic integrity required for survival in an ever-fluctuating chemical environment.
The Biology — How It Works

Magnesium Blend – The Most Important Mineral
A high-bioavailability mineral blend designed to support over 300 essential biochemical reactions, from energy production to muscle relaxation. This formula helps combat daily fatigue while providing the foundational support your nervous system and bones require.
Vetting Notes
Pending
The orchestration of systemic homeostasis is predicated upon the nephron’s capacity to execute high-fidelity molecular discrimination. At the core of this physiological rigour is the regulation of blood pH, maintained within the narrow window of 7.35 to 7.45. This is not merely a passive filtration process but a sophisticated, energy-dependent reclamation and secretion programme. The Proximal Convoluted Tubule (PCT) serves as the primary site for bicarbonate ($HCO_3^-$) reclamation, where approximately 80–90% of the filtered load is recovered. This mechanism is catalysed by Carbonic Anhydrase (CA) isoforms—specifically luminal CA-IV and cytoplasmic CA-II. As research published in *The Lancet* underscores, the failure of this proximal reclamation leads to Type 2 Renal Tubular Acidosis, demonstrating the absolute necessity of the $Na^+/H^+$ exchanger (NHE3) in the apical membrane. By secreting $H^+$ into the lumen, the nephron facilitates the conversion of filtered $HCO_3^-$ into $CO_2$ and $H_2O$, which then diffuse into the tubular cell to be reconstituted into bicarbonate and exported across the basolateral membrane via the $Na^+/HCO_3^-$ cotransporter (NBCe1).
Beyond mere reclamation, the nephron must generate 'new' bicarbonate to offset the non-volatile acid load produced by protein metabolism. This is achieved through ammoniagenesis, a process primarily localised in the PCT where glutamine is metabolised into ammonium ($NH_4^+$) and alpha-ketoglutarate. The subsequent excretion of $NH_4^+$ into the tubular lumen—coupled with the return of de novo $HCO_3^-$ to the systemic circulation—represents a critical "truth" of renal physiology: the kidney does not just filter; it actively synthesises the chemical buffers required for survival. This biological imperative is further refined in the distal segments, specifically within the Intercalated Cells (ICs) of the collecting duct. Type A ICs utilise $H^+$-ATPase and $H^+/K^+$-ATPase pumps to secrete protons against a steep electrochemical gradient, a process vital for the excretion of titratable acidity, primarily as dihydrogen phosphate ($H_2PO_4^-$).
Simultaneously, the nephron governs electrolyte stoichiometry through the countercurrent multiplier system in the Loop of Henle and the fine-tuning mechanisms of the Distal Convoluted Tubule (DCT). The Thick Ascending Limb (TAL) employs the $Na^+$-$K^+$-$2Cl^-$ cotransporter (NKCC2) to reabsorb sodium, creating the medullary osmotic gradient essential for water conservation. At INNERSTANDIN, we recognise that the precision of these transporters is the vanguard against hypertensive pathologies. Clinical data from PubMed-indexed cohorts in the UK highlights that dysregulation in the Epithelial Sodium Channel (ENaC) in the collecting duct—often driven by aldosterone-mediated signaling—is a primary driver of refractory hypertension. The nephron’s ability to balance $K^+$ secretion via the Renal Outer Medullary Potassium (ROMK) channel while simultaneously reclaiming $Na^+$ demonstrates a level of metabolic precision that ensures extracellular fluid volume and electrical excitability remain stable. This intricate dance of ions and protons is the fundamental biological basis for systemic resilience, proving that the nephron is the ultimate arbiter of the organism’s internal environment.
Mechanisms at the Cellular Level
The fundamental architecture of renal regulation is predicated upon a sophisticated suite of transcellular and paracellular transport mechanisms, orchestrated by the electrochemical gradients established by the basolateral Na+/K+-ATPase pump. To truly INNERSTANDIN the nephron is to perceive it as a biological computer, processing vast quantities of plasma filtrate to maintain a narrow physiological pH of 7.35–7.45. This precision is most evident within the Proximal Convoluted Tubule (PCT), where approximately 80% of filtered bicarbonate (HCO3−) is reclaimed. This is not a simple diffusion process; it is an enzymatic cycle facilitated by Carbonic Anhydrase (CA) isoforms. Apical CA-IV dehydrates luminal HCO3− into CO2 and H2O, allowing for rapid intracellular diffusion. Once internalised, cytoplasmic CA-II rehydrates these components back into HCO3− and H+, with the former being exported across the basolateral membrane via the sodium-bicarbonate cotransporter NBCe1 (SLC4A4). Research published in *Kidney International* highlights that mutations in the SLC4A4 gene lead to proximal renal tubular acidosis, underscoring the non-negotiable nature of this cellular pathway.
Beyond the PCT, the distal segments of the nephron—specifically the Distal Convoluted Tubule (DCT) and the Collecting Duct (CD)—serve as the site of ultimate 'fine-tuning' under hormonal governance. Here, the cellular landscape diverges into specialised populations: Principal cells and Intercalated cells. Principal cells are the primary site for sodium (Na+) reabsorption and potassium (K+) secretion, driven largely by the epithelial sodium channel (ENaC) and the renal outer medullary potassium (ROMK) channel. This exchange is heavily influenced by aldosterone, which upregulates channel expression to preserve extracellular fluid volume. However, the true complexity of acid-base homeostasis lies within the Intercalated cells. Type A (alpha) intercalated cells are the protagonists of acid excretion during acidosis, utilizing apical H+-ATPases and H+/K+-ATPases to pump protons into the tubular lumen against steep concentration gradients. Concurrently, they return HCO3− to the systemic circulation via the basolateral anion exchanger 1 (AE1).
Conversely, Type B (beta) intercalated cells, though less numerous, are essential for alkalosis compensation. They exhibit a functional inversion, secreting HCO3− into the urine via the apical Cl−/HCO3− exchanger pendrin (SLC26A4) while sequestering H+ into the blood. This dynamic switching, documented in peer-reviewed studies across *The Lancet* and *Nature Reviews Nephrology*, represents an unparalleled level of biological adaptability. Furthermore, the nephron’s ability to generate 'new' bicarbonate through ammoniagenesis—the breakdown of glutamine in the PCT—provides a secondary buffer system that is vital during chronic metabolic acid stress. By understanding these high-density molecular interactions, we move beyond superficial anatomy to INNERSTANDIN the metabolic truth of renal preservation: a relentless, ATP-dependent struggle to maintain systemic equilibrium against the constant influx of metabolic acids. This cellular precision ensures that the electrolyte concentrations required for neuronal signalling and muscular contraction remain within the requisite haemodynamic tolerances.
Environmental Threats and Biological Disruptors
The delicate homeostatic equanimity maintained by the nephron is increasingly besieged by a clandestine array of exogenous stressors, collectively termed the 'exposome'. While the kidney possesses remarkable compensatory plasticity, modern environmental disruptors exert pathological pressures that bypass traditional evolutionary defences, leading to the insidious erosion of pH and electrolyte regulation. At the vanguard of these threats are heavy metals, most notably Cadmium (Cd) and Lead (Pb), which remain persistent contaminants in UK industrial legacies and certain water distribution infrastructures. Cadmium, sequestered within the renal cortex with a biological half-life exceeding two decades, is notoriously nephrotoxic. Research published in journals such as *The Lancet Planetary Health* highlights that even low-level chronic exposure triggers proximal tubular dysfunction. Mechanistically, Cd enters the proximal tubule cells via receptor-mediated endocytosis, mimicking essential ions. Once intracellular, it induces oxidative stress, inactivating the Na+/K+-ATPase pumps and disrupting the megalin-cubilin transport system. This leads to a profound failure in bicarbonate reabsorption and a subsequent Type 2 Renal Tubular Acidosis, fundamentally destabilising systemic pH.
Furthermore, the ubiquity of per- and polyfluoroalkyl substances (PFAS)—the so-called 'forever chemicals'—presents a burgeoning crisis for renal physiology. In the UK context, monitoring of various water basins has revealed concentrations that may interfere with the organic anion transporters (OATs) located on the basolateral membrane of the nephron. These transporters are critical for the secretion of metabolic waste and the fine-tuning of electrolyte gradients. Evidence suggests that PFAS compete for binding sites, potentially reducing the clearance of endogenous acids and leading to a state of chronic, low-grade metabolic acidosis. This is compounded by the widespread consumption of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) within the British population. NSAIDs inhibit the synthesis of prostaglandins (PGE2 and PGI2), which are vital for maintaining renal blood flow via afferent arteriolar vasodilation. In the absence of these vasodilatory signals, the nephron suffers from transient ischaemia, impairing the juxtaglomerular apparatus's ability to regulate the Renin-Angiotensin-Aldosterone System (RAAS), thereby triggering hyperkalaemia and sodium retention.
At INNERSTANDIN, we must interrogate the impact of the modern dietary landscape, specifically the 'Potential Renal Acid Load' (PRAL) inherent in highly processed, phosphate-rich diets common in urban UK environments. Excessive dietary acid forces the nephron to undergo hypertrophy of the intercalated cells to ramp up ammoniagenesis. While this provides a short-term buffer, the resulting high intrarenal ammonia concentrations activate the alternative complement pathway, inciting tubulointerstitial fibrosis. This transition from physiological adaptation to pathological scarring is the silent precursor to Chronic Kidney Disease (CKD), where the nephron's precision is permanently sacrificed to the altar of environmental and chemical saturation. Such biological disruption necessitates a rigorous re-evaluation of our systemic interactions with these pervasive molecular antagonists.
The Cascade: From Exposure to Disease
The transition from homeostatic precision to pathological decay within the nephron represents a definitive bioenergetic collapse, often initiated by sustained environmental and metabolic insults. This cascade begins with the exhaustion of the proximal convoluted tubule (PCT), an area responsible for the reabsorption of approximately 80% of filtered bicarbonate ($HCO_3^-$). Under the pressure of a high-protein, acid-ash diet—prevalent in the UK and broader Western demographics—the nephron is forced into a state of chronic hyper-filtration to maintain a blood pH of 7.35 to 7.45. Research published in *Nature Reviews Nephrology* indicates that this compensatory mechanism eventually leads to the depletion of intracellular glutamine, the primary substrate for renal ammoniagenesis. When the PCT can no longer generate sufficient ammonia ($NH_3$) to buffer hydrogen ions ($H^+$), the system shifts from physiological regulation to a state of systemic acidosis, triggering a pro-inflammatory cytokine storm that targets the renal interstitium.
At INNERSTANDIN, we recognise that this is not merely a localised failure but a systemic breach. As the PCT fails, the burden of pH regulation shifts to the distal nephron’s intercalated cells. Type A intercalated cells attempt to compensate by upregulating $H^+$-ATPase pumps to extrude excess acid into the tubular lumen. However, this process is energy-intensive and, in the presence of chronic hyper-aldosteronism—often driven by high sodium intake or vascular stiffening—leads to an obligatory loss of potassium ($K^+$). The resulting hypokalaemia induces a state of intracellular acidosis even when extracellular pH appears normal, a phenomenon that masks the underlying cellular distress from standard clinical assays.
The cascade further accelerates through the disruption of the Renin-Angiotensin-Aldosterone System (RAAS). According to longitudinal data in *The Lancet*, the persistent activation of RAAS due to nephronal "over-work" results in glomerular hypertension and the subsequent breakdown of the podocyte slit diaphragm. This protein leakage (proteinuria) is not just a symptom but a primary driver of further disease, as the reabsorption of filtered proteins is toxic to tubular cells, inducing mitochondrial fragmentation and epithelial-to-mesenchymal transition (EMT). Consequently, the kidney’s precision in balancing electrolytes like sodium and calcium is sacrificed to maintain immediate survival, leading to the interstitial fibrosis and tubular atrophy (IFTA) that characterises end-stage renal disease. The "truth-exposing" reality is that the nephron's failure to maintain pH balance is the silent architect of systemic cardiovascular remodelling, as the body pulls minerals from the skeletal system to buffer the acidic blood, illustrating a total loss of biological INNERSTANDIN of the kidney's original regulatory blueprint.
What the Mainstream Narrative Omits
The conventional clinical discourse surrounding renal function frequently reduces the nephron to a mechanical sieve, prioritising Glomerular Filtration Rate (GFR) while overlooking the sophisticated bio-molecular signalling that governs systemic homeostasis. At INNERSTANDIN, we recognise that the mainstream narrative fails to adequately address the metabolic cost of ammoniogenesis and the nuances of the distal tubule’s "intercalated cell" duality. While basic physiology textbooks suggest a binary system of filtration and excretion, the reality is a high-resolution, ATP-dependent orchestration of ionic flux that dictates the very viability of cellular respiration.
The primary omission in common medical literature is the sheer complexity of the kidney’s role in ‘de novo’ bicarbonate synthesis. Standard models focus on bicarbonate reclamation in the proximal convoluted tubule (PCT) via carbonic anhydrase II and IV; however, they often marginalise the significance of glutamine metabolism. In states of chronic metabolic stress—often exacerbated by the high-PRAL (Potential Renal Acid Load) diets prevalent in the UK—the nephron must upregulate the extraction of glutamine from the peritubular capillaries. Research indexed in PubMed highlights that for every molecule of glutamine metabolised in the PCT, two ammonium ions ($NH_4^+$) are excreted and two new bicarbonate ions are generated. This is not merely a compensatory mechanism; it is a fundamental shift in renal substrate priority that, if sustained, leads to tubular hypertrophy and oxidative stress.
Furthermore, the mainstream narrative provides a cursory glance at the Type A and Type B intercalated cells within the cortical collecting duct, yet it ignores the critical role of Pendrin (anion exchanger SLC26A4). While Type A cells utilise V-ATPase pumps to secrete protons into the lumen during acidosis, Type B cells are essential for correcting alkalosis by secreting bicarbonate. The precision of this "switch" is governed by local pH sensors and mineralocorticoid activity, a mechanism that remains under-researched in standard General Practitioner training. Evidence from *The Lancet* and various nephrology journals suggests that even subtle dysregulation in this intercalated cell ratio can lead to "low-grade metabolic acidosis" (LGMA). LGMA is often clinically silent—falling within the so-called "normal" range of 22-29 mmol/L—yet it is potent enough to trigger bone mineral resorption and muscle proteolysis. By focusing only on overt pathological states, the mainstream ignores the sub-clinical renal strain that precedes chronic kidney disease (CKD) and systemic metabolic collapse. At INNERSTANDIN, we posit that the nephron’s precision is not a static state of filtration, but a dynamic, resource-intensive defence against the systemic acid-ash burden of modern life.
The UK Context
In the British clinical landscape, the nephron’s capacity for homeostatic regulation is not merely a textbook abstraction but a critical frontline defence against the systemic metabolic derangements prevalent in the UK population. Data from the UK Renal Registry (UKRR) underscores a burgeoning crisis: approximately 7.2 million people in the UK are living with Chronic Kidney Disease (CKD) stages 3-5, a condition that fundamentally compromises the precision of acid-base and electrolyte titration. At the cellular level, the nephron’s architectural integrity determines the efficacy of the bicarbonate ($HCO_3^-$) buffering system. Under physiological norms, the proximal convoluted tubule (PCT) reabsorbs roughly 80% of filtered bicarbonate via the apical $Na^+/H^+$ exchanger 3 (NHE3) and the basolateral $Na^+/HCO_3^-$ cotransporter (NBCe1). However, INNERSTANDIN research highlights that the typical British diet—characterised by a high intake of processed proteins and cereal grains—induces a chronic net acid load (NAL), forcing the nephron into a state of perpetual compensatory ammoniagenesis.
The precision of the distal nephron, specifically the Type A intercalated cells of the collecting duct, becomes the final arbiter of blood pH. Through the active secretion of protons via $H^+$-ATPase and $H^+/K^+$-ATPase pumps, the nephron facilitates the de novo synthesis of bicarbonate. Yet, longitudinal analyses within the UK Biobank cohort reveal that even minor decrements in glomerular filtration rate (GFR) lead to the retention of fixed acids, precipitating sub-clinical metabolic acidosis. This is not a benign state; according to research published in *The Lancet*, persistent acidotic stress triggers a maladaptive intra-renal RAAS activation, accelerating fibrotic pathways. Furthermore, the UK’s specific demographic challenges, including a high prevalence of hypertension, exacerbate the pressure-natriuresis relationship. The nephron's inability to precisely modulate sodium-potassium exchange via the ENaC channels in the distal tubule leads to systemic potassium shifts, which, as evidenced by PubMed-indexed studies on UK primary care patients, increases the risk of cardiac arrhythmias. This systemic failure highlights that the nephron is the metabolic fulcrum upon which British public health teeters; its precision is the only barrier between physiological stability and the catastrophic collapse of internal milieu.
Protective Measures and Recovery Protocols
The intrinsic resilience of the nephron is predicated upon a sophisticated hierarchy of autoregulatory feedbacks and cellular salvage pathways designed to mitigate the deleterious effects of metabolic acidosis and electrolyte flux. Central to these protective measures is the tubuloglomerular feedback (TGF) mechanism, orchestrated by the macula densa within the juxtaglomerular apparatus. When the nephron detects an elevation in luminal sodium chloride—indicative of potential hyperfiltration or proximal dysfunction—it triggers a paracrine signalling cascade involving adenosine and ATP. This induces vasoconstriction of the afferent arteriole, thereby modulating the glomerular filtration rate (GFR) to prevent distal solute overload and preserve the delicate haemodynamic balance within the vasa recta. At INNERSTANDIN, we recognise that this is not merely a regulatory loop but a fundamental survival protocol that shields the medulla from the hypoxic stress associated with excessive active transport.
In the context of acid-base homeostasis, the nephron employs an exhaustive recovery protocol through the induction of ammoniagenesis, primarily within the S3 segment of the proximal tubule. Research published in *The Lancet* and *Nature Reviews Nephrology* highlights that during chronic metabolic acidosis, the kidney undergoes structural hypertrophy to increase its capacity for glutamine extraction. By deaminating glutamine, the nephron generates "new" bicarbonate (HCO3−) ions whilst simultaneously producing ammonium (NH4+), which serves as a critical urinary buffer. This systemic restoration is facilitated by the up-regulation of the SNAT3 transporter and phosphate-dependent glutaminase (PDG), ensuring that the plasma pH remains within the narrow physiological window of 7.35 to 7.45. This molecular repositioning is a testament to the nephron's capacity for plastic adaptation in the face of systemic insult.
Furthermore, the recovery of the nephron following ischaemic or toxic injury—often categorised as Acute Kidney Injury (AKI) in UK clinical settings—relies on the dedifferentiation and proliferation of surviving tubular epithelial cells. Unlike many other mammalian tissues, the proximal tubule possesses a remarkable, albeit finite, regenerative capacity. During the recovery phase, cells express mesenchymal markers such as Vimentin and SOX9, reverting to a progenitor-like state to repopulate the basement membrane. However, INNERSTANDIN research underscores a "truth-exposing" caveat: if the inflammatory milieu persists, this recovery protocol can become maladaptive, leading to the activation of the myofibroblast transition and subsequent interstitial fibrosis. To counter this, the nephron utilises endogenous antioxidant defences, such as the Nrf2-Keap1 pathway, which up-regulates cytoprotective enzymes like haeme oxygenase-1 (HO-1) to neutralise reactive oxygen species (ROS) generated during reperfusion. This intricate interplay between immediate haemodynamic shielding and long-term cellular regeneration constitutes the nephron's primary defence against the dissolution of systemic homeostasis.
Summary: Key Takeaways
The renal nephron operates as the quintessential nexus for systemic physiological stability, orchestrating an intricate balance between metabolic exigencies and haemodynamic pressures. Evidence corroborated by research published in *The Lancet* and various PubMed-indexed longitudinal studies underscores that the kidneys do not merely filter waste; they execute a high-precision molecular audit of blood chemistry. Central to this is the proximal convoluted tubule’s reclamation of approximately 80–90% of filtered bicarbonate ($HCO_3^-$), a process facilitated by membrane-bound and cytosolic carbonic anhydrase isoforms that are fundamental to preventing systemic metabolic acidosis. At INNERSTANDIN, we recognise that the distal nephron’s Type A and Type B intercalated cells further refine this pH through the ATP-driven secretion of hydrogen ions and the synthesis of *de novo* bicarbonate via ammoniagenesis, a mechanism that remains the body's primary defence against chronic acid loading.
Furthermore, electrolyte regulation—governed by the renin-angiotensin-aldosterone system (RAAS) and the exquisite sensitivity of the distal convoluted tubule to mineralocorticoids—dictates cardiovascular tone, extracellular fluid volume, and neuronal excitability. The precision of the countercurrent multiplier system within the Loop of Henle ensures osmotic gradients are maintained with mathematical rigour, a process UK-based clinical research suggests is critical for preventing the long-term sequelae of electrolyte-induced cardiac arrhythmias. Ultimately, the nephron’s capacity to maintain this delicate equilibrium represents a masterclass in biological engineering, where minute deviations in tubular transport protein expression can precipitate catastrophic systemic failure, necessitating a profound INNERSTANDIN of these micro-physiological drivers.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "The Nephron's Precision: How the Kidneys Regulate Blood pH and Electrolyte Balance"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Physiology — products curated by our research team for educational relevance and biological support.
Magnesium Blend – The Most Important Mineral

Magnesium Spray – Easy-to-Use Topical Magnesium

Fulvic Minerals – Natural Rare Earth Minerals. The essential trace elements missing from modern processed foods.
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.