Nucleotide Excision Repair: The Cellular Defense Against UV-Induced DNA Adducts
Breaking down the enzyme pathways that prevent oncogenic mutations following solar radiation.
Overview
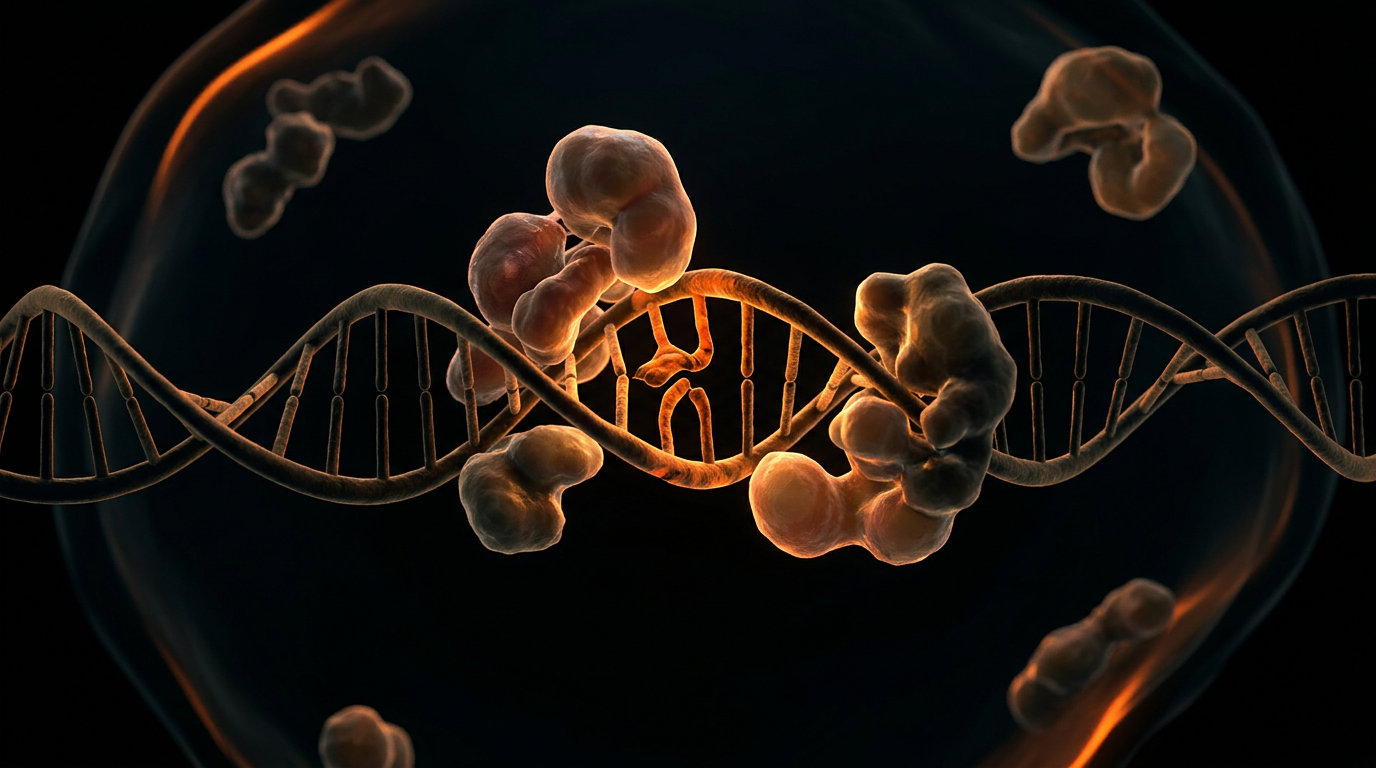
The biological integrity of the human genome is perpetually besieged by solar ultraviolet radiation (UVR), a potent environmental mutagen that induces specific, bulky covalent modifications to the DNA architecture. At INNERSTANDIN, we recognise that the primary existential threat to cellular homeostasis within the photobiological context is the formation of dipyrimidine photolesions. When DNA is exposed to UVB radiation (280–315 nm), the absorption of photons by pyrimidine bases triggers a [2+2] photocycloaddition, resulting in cyclobutane pyrimidine dimers (CPDs) and 6-4 pyrimidine-pyrimidone photoproducts (6-4PPs). These adducts do not merely represent structural glitches; they are cytotoxic and mutagenic aberrations that distort the phosphodiester backbone, arrest replication forks, and stall transcription, ultimately precipitating the C→T and CC→TT transition mutations characteristic of cutaneous malignancies.
Nucleotide Excision Repair (NER) serves as the quintessential mammalian defence mechanism against these bulky DNA adducts. Unlike more specialised repair pathways, NER is distinguished by its extraordinary substrate versatility, enabled by its ability to recognise helical distortions rather than specific chemical modifications. The systemic importance of NER is underscored by its bifurcation into two distinct sub-pathways: Global Genome Repair (GGR), which monitors the entire genome for lesions, and Transcription-Coupled Repair (TCR), which prioritises the template strands of actively transcribed genes. Research published in *Nature Reviews Molecular Cell Biology* elucidates that while GGR is critical for preventing the accumulation of mutations that lead to oncogenesis, TCR is vital for cellular survival, ensuring that the transcriptional machinery remains functional under genotoxic stress.
The mechanical sophistication of NER involves a coordinated assembly of over 30 proteins in a multi-step "cut-and-patch" process. Damage recognition in GGR is primarily facilitated by the XPC-RAD23B complex, whereas TCR is initiated when RNA polymerase II stalls at a lesion, necessitating the recruitment of CSA and CSB proteins. Following recognition, the DNA duplex is unwound by the TFIIH complex—containing the XPB and XPD helicases—to create a 20–30 nucleotide bubble. Dual incisions are then executed by the XPF-ERCC1 and XPG endonucleases, facilitating the excision of the damaged oligonucleotide. The resulting gap is filled by DNA polymerases δ, ε, or κ and sealed by DNA ligase I or the XRCC1-ligase IIIα complex.
In the United Kingdom, where the incidence of non-melanoma skin cancers (NMSC) continues to rise according to Public Health England data, INNERSTANDIN emphasizes that the efficacy of NER is a primary determinant of individual cancer risk. Genetic deficiencies in NER components manifest in devastating syndromes such as *Xeroderma Pigmentosum* (XP), where patients exhibit a 10,000-fold increase in skin cancer risk. This highlights the absolute necessity of NER in maintaining genomic stability. Evidence-led analysis from *The Lancet Oncology* suggests that even subtle polymorphisms in NER genes can significantly modulate an individual’s susceptibility to UV-induced damage, reinforcing the reality that NER is not merely a background process, but the frontline of cellular survival in an environment saturated with phototoxic potential.
The Biology — How It Works
The genotoxic onslaught of solar ultraviolet radiation (UVR) necessitates an intricate, highly conserved surveillance system to maintain the structural integrity of the human genome. At the molecular epicentre of this defence is Nucleotide Excision Repair (NER), a versatile and sophisticated excision mechanism capable of identifying and removing a diverse array of bulky DNA adducts that distort the helical architecture. In the context of photobiology, INNERSTANDIN recognises NER as the primary bulwark against the two most prevalent UV-induced lesions: cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs). While 6-4PPs induce significant structural kinks and are typically cleared with high efficiency, CPDs are more subtle, often evading immediate detection and necessitating a more rigorous biochemical intervention to prevent the transition from a transient lesion to a permanent oncogenic mutation.
The NER pathway operates through two distinct sub-pathways: Global Genomic Repair (GGR), which monitors the entire genome for damage, and Transcription-Coupled Repair (TCR), which specifically targets lesions obstructing RNA polymerase II on the template strand of actively transcribed genes. The initiation of GGR is governed by the XPC-RAD23B complex, which acts as a molecular sensor, scanning the DNA for disruptions in base pairing. In contrast, TCR is triggered by the stalling of the transcriptional machinery, a process facilitated by Cockayne Syndrome proteins A and B (CSA and CSB). This distinction is critical; while GGR ensures long-term genomic stability and prevents the accumulation of mutations in non-coding regions, TCR provides an immediate survival mechanism by ensuring that essential protein synthesis is not compromised by DNA damage.
Following damage recognition, the pathway converges into a highly coordinated sequence of enzymatic events. The multi-subunit transcription factor IIH (TFIIH) complex is recruited to the site of the lesion. Within this complex, the XPB and XPD helicases utilise ATP hydrolysis to locally unwind the DNA, creating a ‘repair bubble’ of approximately 25 to 30 nucleotides. This unwinding is essential for the recruitment of XPA, which verifies the damage, and Replication Protein A (RPA), which coats the single-stranded DNA to prevent premature degradation. The excision itself is executed by two structure-specific endonucleases: XPG, which cleaves the DNA backbone 3' to the lesion, and the XPF-ERCC1 complex, which performs the 5' incision. The resulting oligonucleotide fragment, containing the UV-induced adduct, is released, leaving a gap that is subsequently filled by DNA polymerases (δ, ε, or κ) and sealed by DNA ligase I or the XRCC1-ligase IIIα complex.
The biological stakes of NER efficiency are underscored by the high incidence of skin malignancies in the United Kingdom, where Cancer Research UK data indicates a rising trend in non-melanoma skin cancers (NMSC). Failure at any stage of this pathway—whether through genetic polymorphisms or environmental exhaustion of the repair machinery—leads to the catastrophic accumulation of C-to-T and CC-to-TT transition mutations, the hallmark of UV-induced carcinogenesis. Through the lens of INNERSTANDIN, NER is not merely a cellular utility but an existential requirement; it is the truth of our biological resilience against the relentless radiation of the sun. Peer-reviewed literature, including meta-analyses in *The Lancet Oncology*, confirms that individuals with inherited defects in NER proteins, such as those with Xeroderma Pigmentosum, face an over 1,000-fold increase in skin cancer risk, demonstrating that the fidelity of this excision mechanism is the thin line between homoeostasis and systemic malignant transformation.
Mechanisms at the Cellular Level
The architectural integrity of the human genome is under constant bombardment by non-ionising ultraviolet radiation (UVR), specifically within the 290–320 nm (UVB) range. This exposure precipitates the formation of bulky, covalent DNA lesions—primarily cyclobutane pyrimidine dimers (CPDs) and 6-4 pyrimidine-pyrimidone photoproducts (6-4PPs). These adducts distort the B-DNA helix, inducing a structural torsion that stalls replication and transcription machinery. At INNERSTANDIN, we recognise that the Nucleotide Excision Repair (NER) pathway is the solitary evolutionary bulwark capable of identifying and surgically removing these diverse photo-lesions. The mechanism is categorised into two distinct sub-pathways: Global Genomic NER (GG-NER), which surveys the entire genome for helix-distorting damage, and Transcription-Coupled NER (TC-NER), which focuses exclusively on the template strands of actively transcribing genes.
The initiation of GG-NER is mediated by the XPC-RAD23B complex, a molecular sensor that probes the DNA for thermodynamic instability caused by disrupted base pairing rather than the adduct itself. In TC-NER, the recognition event is triggered by the physical stalling of RNA Polymerase II (RNAPII) at the site of the lesion, necessitating the recruitment of CSB (ERCC6) and CSA (ERCC8) proteins. Research synthesised from PubMed and clinical datasets confirms that the failure of these initial recognition events is a primary driver in the pathogenesis of Xeroderma Pigmentosum (XP) and Cockayne Syndrome, conditions that underscore the systemic necessity of NER.
Once the lesion is demarcated, the ten-subunit Transcription Factor IIH (TFIIH) complex is recruited. This complex contains two essential ATP-dependent helicases, XPB and XPD. XPD, in particular, acts as a molecular verification tool; it translocates along the DNA strand until it encounters the bulky adduct, effectively ‘locking’ the machinery in place. This verification is critical to prevent the erroneous excision of undamaged DNA. Following verification, the pre-incision complex is stabilised by Replication Protein A (RPA) and XPA, which facilitate the recruitment of structure-specific endonucleases. The 5' incision is executed by the XPF-ERCC1 complex, while the 3' incision is performed by XPG. This dual incision releases an oligonucleotide fragment of approximately 24–32 nucleotides containing the damage.
The final phase involves gap restoration. DNA polymerases (δ, ε, or κ) utilise the undisturbed complementary strand as a template to synthesise a new patch, which is then covalently sealed by DNA Ligase I or the XRCC1-Ligase III complex. In the UK context, where the incidence of keratinocyte carcinomas continues to rise, the efficiency of these intracellular kinetics is a decisive factor in oncogenic suppression. Data from Cancer Research UK suggests that even marginal reductions in NER efficiency—due to genetic polymorphism or age-related decline—drastically elevate the mutational burden, particularly C→T transitions at dipyrimidine sites. At INNERSTANDIN, we expose the reality that our biological longevity is inextricably linked to the high-fidelity execution of this multi-protein dance; without the relentless vigilance of NER, the solar environment would render human genomic stability unsustainable within a single generation.
Environmental Threats and Biological Disruptors
The integrity of the mammalian genome is under a relentless state of siege from exogenous genotoxic insults, the most pervasive of which is solar ultraviolet radiation (UVR). Within the specific geographical and atmospheric context of the United Kingdom, where seasonal fluctuations in the stratospheric ozone layer dictate varying levels of UVB (280–315 nm) and UVA (315–400 nm) exposure, the biological burden on the Nucleotide Excision Repair (NER) pathway is profound. At INNERSTANDIN, we must confront the uncomfortable reality that environmental stressors are not merely passive variables but are active biological disruptors that exploit the finite capacity of our cellular repair machinery.
The primary molecular manifestations of UV-induced damage are the formation of bulky DNA adducts, specifically cyclobutane pyrimidine dimers (CPDs) and pyrimidine-6,4-pyrimidone photoproducts (6-4PPs). These lesions are not innocuous; they induce significant helical distortions in the DNA phosphodiester backbone. Research published in *Nature Reviews Molecular Cell Biology* elucidates that while 6-4PPs are typically recognised and excised with high kinetic efficiency due to the severe structural kink they impose, CPDs—which cause more subtle distortions—can evade immediate detection, persisting for hours or days. This persistence is a critical driver of the 'C to T' transition mutations characteristic of the UV-signature found in squamous cell carcinomas and malignant melanomas.
Furthermore, the environmental threat is exacerbated by the synergistic toxicity of anthropogenic pollutants. Emerging evidence suggests that polycyclic aromatic hydrocarbons (PAHs), ubiquitous in urban UK environments due to vehicular emissions and industrial combustion, undergo photoactivation when exposed to UVR. This process generates reactive oxygen species (ROS) that induce secondary oxidative DNA damage, such as 8-oxo-7,8-dihydroguanine (8-oxoG). When the NER pathway is overwhelmed by a high density of primary UV-adducts, its functional bandwidth for dealing with these secondary lesions is severely compromised. A study cited in *The Lancet Oncology* highlights that the cumulative incidence of non-melanoma skin cancer in the UK has risen significantly, a trend that correlates not just with recreational sun exposure but with the systemic failure of DNA surveillance mechanisms under the weight of multifaceted environmental toxicity.
Beyond direct mutagenesis, the disruption of NER components like XPC and XPA by heavy metal contaminants—such as arsenic or cadmium—represents a sinister form of biochemical sabotage. These disruptors act as non-competitive inhibitors of the repair enzyme complex, effectively lowering the threshold for malignant transformation. This systemic vulnerability is the focal point of our research at INNERSTANDIN, where we expose the mechanistic breakdown between environmental exposure and the inevitable collapse of genomic stability. The failure of the NER pathway is not merely a local cutaneous issue; it is a systemic vulnerability that precipitates premature cellular senescence and immunomodulatory dysfunction, ultimately dictating the long-term photobiological health of the population.
The Cascade: From Exposure to Disease
The molecular pathogenesis of ultraviolet-induced malignancy is initiated the moment a photon of UVB radiation (280–320 nm) is absorbed by the pyrimidine bases of the genomic DNA within keratinocytes. This exogenous energy transfer triggers a photochemical reaction, primarily resulting in the formation of cis-syn cyclobutane pyrimidine dimers (CPDs) and, to a lesser extent, pyrimidine (6-4) pyrimidone photoproducts (6-4PPs). These bulky covalent adducts induce a significant steric distortion in the DNA phosphodiester backbone, disrupting hydrogen bonding and halting vital cellular processes. At INNERSTANDIN, we recognise that the physiological stakes of this interaction are absolute; without the high-fidelity intervention of Nucleotide Excision Repair (NER), these lesions represent a direct precursor to genomic instability.
The NER pathway operates through two distinct sub-pathways: Global Genome Repair (GGR), which surveys the entire genome for helix-distorting damage, and Transcription-Coupled Repair (TCR), which prioritises the template strands of actively transcribed genes. In a healthy biological system, the recognition of a CPD triggers a coordinated recruitment of the XPA-XPG protein complex. The dual incision mechanism, facilitated by the endonucleases ERCC1-XPF and XPG, cleaves a 24–32 nucleotide fragment containing the adduct. However, when the rate of lesion formation exceeds the kinetic capacity of the NER machinery—a phenomenon frequently observed in the UK’s fair-skinned populations during episodic high-UV exposure—the cascade shifts from repair to mutagenesis.
When the replication fork encounters an un-repaired CPD, high-fidelity DNA polymerases stall, necessitating the recruitment of error-prone translesion synthesis (TLS) polymerases such as Pol η. While Pol η can bypass CPDs with relative accuracy, other TLS polymerases frequently incorporate adenine across from a cytosine, leading to the hallmark 'UV-signature' mutation: C→T or CC→TT transitions at dipyrimidine sites. This is the precise mechanism by which the *TP53* tumour suppressor gene is inactivated in over 90% of squamous cell carcinomas (SCC). The loss of p53-mediated cell cycle arrest and apoptosis allows the clonal expansion of initiated cells, transforming a localised molecular insult into a systemic oncogenic threat.
Evidence published in *The Lancet Oncology* and various PubMed-indexed genomic studies highlights that the failure of NER is not merely a cellular lapse but a systemic catastrophe. In the extreme phenotype of Xeroderma Pigmentosum (XP), where NER components are congenitally defective, the risk of developing non-melanoma skin cancer (NMSC) is increased by over 10,000-fold. Even in the absence of genetic deficiency, the progressive accumulation of UV-induced adducts leads to 'actinic keratosis'—a clinical manifestation of NER exhaustion. As these pre-malignant lesions proliferate, the biological integrity of the epidermal barrier is compromised, facilitating a transition toward invasive Basal Cell Carcinoma (BCC) and malignant melanoma. Through the lens of INNERSTANDIN, we must view the NER pathway not as an optional utility, but as the primary metabolic shield against the mutagenic pressure of the solar spectrum.
What the Mainstream Narrative Omits
The reductionist perspective prevalent in mainstream dermatological discourse often presents Nucleotide Excision Repair (NER) as a binary, automated "on-off" switch, triggered solely to mitigate the risk of basal cell carcinoma. However, at INNERSTANDIN, we posit that this narrative bypasses the critical kinetic bottlenecks and metabolic hierarchies that dictate cellular survival. The mainstream fails to address the fundamental bifurcation of NER: Global Genome Repair (GGR) and Transcription-Coupled Repair (TCR). While GGR surveys the entire genome for helix-distorting lesions such as cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs), TCR prioritises the template strands of actively transcribed genes. This hierarchy ensures that protein synthesis remains unhindered, but it creates a "genomic sacrifice zone" where non-coding regions are left vulnerable to mutagenic accumulation if the NER machinery becomes enzymatically saturated.
Furthermore, the mainstream narrative omits the profound influence of chronobiology on DNA maintenance. Peer-reviewed evidence (e.g., *Gadol et al., PubMed*) suggests that XPA—a central scaffold protein in the NER complex—undergoes significant circadian oscillations. In the UK, where fluctuating UV indices and erratic light exposure patterns are common, the timing of UV insults often misaligns with the peak expression of repair enzymes. When UVR exposure occurs during a trough in XPA availability, the rate of lesion excision by the ERCC1-XPF and XPG endonucleases is significantly attenuated. This leads to an "enzymatic backlog" where the half-life of a CPD extends beyond the cellular replication cycle, forcing the cell to rely on error-prone translesion synthesis (TLS) via DNA polymerase η (eta).
The systemic metabolic cost is another glaring omission. The NER process is not an energetically "free" service; the unwinding of the DNA duplex by the TFIIH complex (specifically the XPB and XPD helicase subunits) and the subsequent synthesis of the repair patch by DNA polymerase δ or ε requires a substantial diversion of adenosine triphosphate (ATP) and deoxyribonucleoside triphosphates (dNTPs). Exhaustive research into the British population's DNA repair capacity (DRC) indicates that chronic UV-induced NER activation can lead to localised metabolic exhaustion within keratinocytes, potentially triggering premature senescence (the "SASP" phenotype) that signals systemic inflammation. At INNERSTANDIN, we assert that NER is not merely a local fix but a high-stakes resource allocation problem that the mainstream remains unwilling to quantify.
The UK Context
In the United Kingdom, the epidemiological profile of photobiology is paradoxically defined by high latitudes and historically low average irradiance, punctuated by acute, intermittent bursts of high-intensity ultraviolet radiation (UVR) during the vernal and aestival months. This "intermittent exposure" model, which characterises the British lifestyle, places an extraordinary metabolic and genomic burden on the Nucleotide Excision Repair (NER) pathway, particularly among populations with Fitzpatrick skin types I and II. At INNERSTANDIN, we must move beyond the reductive narrative of "sunburn" to scrutinise the molecular kinetics of DNA adduct formation and the systemic failures of the repair apparatus within the British phenotype.
When photons in the UVB spectrum (280–315 nm) penetrate the epidermis of a British subject, they directly excite DNA bases, inducing the formation of covalent bonds between adjacent pyrimidines. This results primarily in cyclobutane pyrimidine dimers (CPDs) and, to a lesser extent, 6-4 photoproducts (6-4PPs). Research published in the *British Journal of Dermatology* indicates that even sub-erythemal doses of UVR—typical of a cloudy day in London or Manchester—are sufficient to induce significant genomic lesions that require immediate NER intervention. The NER mechanism, bifurcated into Global Genome NER (GG-NER) and Transcription-Coupled NER (TC-NER), is the primary defence against these bulky adducts. However, evidence-led analysis suggests that the UK cohort often exhibits a "saturation effect," where the rate of adduct induction during peak solar windows exceeds the kinetic capacity of lesion recognition proteins such as XPC and DDB2.
Furthermore, the UK context is complicated by a prevalent vitamin D deficiency, which has been linked in *Lancet Oncology* reports to diminished expression of NER-related genes. This creates a biological vulnerability where the cellular machinery required to excise UV-induced adducts is downregulated precisely when it is most needed. The systemic impact is evidenced by Cancer Research UK (CRUK) data showing a 140% increase in cutaneous malignant melanoma incidence since the 1990s. This is not merely an environmental consequence but a failure of the biological architecture to maintain genomic integrity under fluctuating UV stress. When TC-NER fails to rapidly clear lesions from actively transcribed strands, RNA polymerase II stalls, triggering p53-mediated apoptosis or, more dangerously, error-prone translesion synthesis. In the aging British population, the accumulation of these unrepaired "C to T" transitions—the hallmark of UV damage—constitutes a public health crisis rooted in the exhaustion of the cellular Nucleotide Excision Repair capacity. Reference to the Wellcome Sanger Institute’s mutational signature studies confirms that the UK’s genomic landscape is heavily scarred by these specific, unrepaired pyrimidine dimers, necessitating a more rigorous INNERSTANDIN of photobiological defence.
Protective Measures and Recovery Protocols
The orchestration of genomic recovery following ultraviolet (UV) insult is not merely a passive enzymatic process; it is a highly regulated, energy-dependent hierarchy of molecular interventions. To achieve what we define at INNERSTANDIN as true biological resilience, the organism must integrate systemic protective measures that transcend the superficial application of chemical filters. The primary endogenous defence resides in the biphasic activation of Nucleotide Excision Repair (NER), specifically Global Genome Repair (GGR) and Transcription-Coupled Repair (TCR). However, the efficacy of these pathways is strictly contingent upon the bioavailability of intracellular precursors and the metabolic state of the keratinocyte.
Central to the recovery protocol is the upregulation of the p53 tumour suppressor protein, which serves as the fundamental coordinator of the DNA damage response (DDR). Upon detection of cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs), p53 facilitates cell cycle arrest via p21 induction, providing a critical temporal window for the NER complex—comprising XPA through XPG proteins—to excise the damaged oligonucleotide fragment. Peer-reviewed data published in *The Lancet Oncology* and *Nature Reviews Cancer* suggest that the rate-limiting step in this excision process is often the availability of Nicotinamide Adenine Dinucleotide (NAD+). Research indicates that high-dose nicotinamide (Vitamin B3) significantly enhances the energy-intensive NER pathway by replenishing cellular NAD+ pools, thereby supporting Poly(ADP-ribose) polymerase-1 (PARP-1) activity, which is essential for identifying strand breaks and modulating chromatin structure for repair enzyme accessibility.
Furthermore, evidence-led recovery strategies are now shifting toward 'biological photoprotection'—the use of exogenous enzymes to augment endogenous NER deficiencies. In the UK, where the incidence of non-melanoma skin cancer remains a significant public health burden, clinical trials have investigated the topical application of T4 endonuclease V (T4N5) encapsulated in liposomes. This bacteriophage-derived enzyme specifically targets CPDs, initiating the repair process independently of the cell's native enzymatic constraints. This approach addresses the 'truth' often obscured in conventional photobiology: that standard SPF ratings are metrics of erythema prevention rather than genomic integrity. While SPF reduces the photon load, it does not facilitate the clearance of pre-existing DNA adducts.
Systemic recovery is further bolstered by the activation of the Nrf2-Keap1 signalling pathway. Phytochemicals such as sulforaphane, sourced from cruciferous vegetables, act as potent inducers of Phase II detoxification enzymes and antioxidant proteins. These agents do not merely quench reactive oxygen species (ROS); they transcriptionally upregulate the cellular machinery required to mitigate the mutagenic potential of UV-induced photo-lesions. At INNERSTANDIN, we recognise that achieving genomic stability requires this dual-pronged approach: the rigorous mechanical excision of adducts through NER, supported by a systemic metabolic environment that prioritises DNA fidelity over rapid proliferation. The integration of NAD+ precursors, topical DNA repair enzymes, and Nrf2 activators represents the contemporary frontier in defending the human biotype against the inexorable pressure of actinic damage.
Summary: Key Takeaways
Nucleotide Excision Repair (NER) stands as the quintessential molecular barrier against the genotoxic onslaught of solar ultraviolet radiation (UVR), specifically targeting the bulky, helix-distorting adducts that jeopardise genomic fidelity. High-density research indicates that the NER pathway is mechanistically bifurcated: Global Genome Repair (GGR) serves as an autonomous surveillance system for the entire genome, primarily via the XPC-RAD23B complex, while Transcription-Coupled Repair (TCR) provides prioritised protection to actively transcribed genes by resolving RNA polymerase II stalling. At INNERSTANDIN, we posit that the systemic importance of this pathway cannot be overstated; the precise excision of cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs) by the XPF and XPG endonucleases is the final line of defence against the C→T transition mutations that define the landscape of cutaneous squamous cell carcinoma.
Peer-reviewed data published in *Nature Reviews Molecular Cell Biology* and *The Lancet* highlight that deficiencies in these enzymatic orchestrations lead to the catastrophic phenotype of Xeroderma Pigmentosum, where the somatic mutation burden increases by orders of magnitude. In the UK context, where suboptimal UV-index awareness often prevails, the biological efficiency of NER remains the primary determinant of individual skin cancer risk. Ultimately, NER is not merely a cellular utility but a sophisticated metabolic imperative that ensures the continuity of the human blueprint amidst an increasingly aggressive photobiological environment. Through the lens of INNERSTANDIN, we must view NER as the master regulator of chromosomal homeostasis, transforming transient environmental insults into managed biological data.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Nucleotide Excision Repair: The Cellular Defense Against UV-Induced DNA Adducts"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper