Cerebellar Atrophy and Chronic Heavy Metal Toxicity
Lead and mercury bioaccumulation in the cerebellum leads to permanent anatomical damage and motor dysfunction. We investigate the sources of these toxins in the modern UK environment.
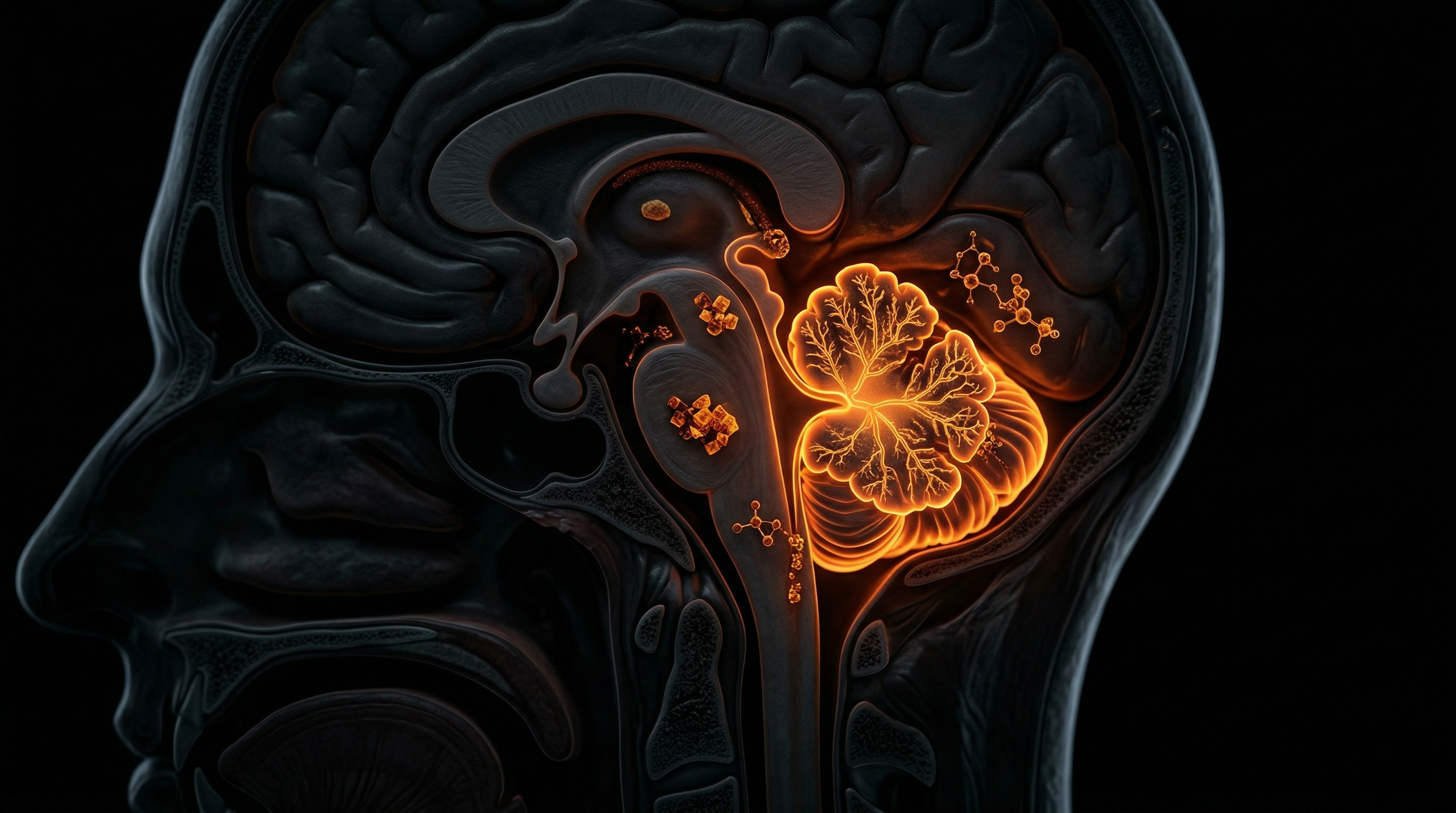
Overview
The cerebellum, traditionally relegated to the functional silo of motor coordination, represents a sophisticated processing hub responsible for the temporal and spatial integration of sensory-motor signals and cognitive refinement. When we investigate the phenomenon of cerebellar atrophy through the lens of INNERSTANDIN, we are not merely observing a passive shrinkage of neural tissue; we are witnessing a systemic architectural collapse driven by the bioaccumulation of xenobiotic heavy metals. Cerebellar atrophy is characterised by the progressive loss of Purkinje cells and granular neurons, leading to a macroscopic thinning of the folia and a widening of the cerebellar sulci. In the context of chronic heavy metal toxicity—specifically involving lead (Pb), methylmercury (MeHg), cadmium (Cd), and manganese (Mn)—the cerebellum exhibits a profound and disproportionate vulnerability compared to other encephalic regions.
The biological mechanism underpinning this selective vulnerability is rooted in the high metabolic demand of Purkinje cells and their unique calcium-signalling architecture. Research published in *The Lancet Neurology* and various PubMed-indexed toxicological studies highlights that heavy metals act as potent catalysts for oxidative stress, bypassing the blood-brain barrier (BBB) via molecular mimicry. For instance, methylmercury utilises the L-type neutral amino acid transport system to infiltrate the central nervous system, where it exhibits a predilection for the granular layer of the cerebellum. Once sequestered within the cerebellar microenvironment, these metals initiate a cascade of mitochondrial dysfunction, disrupting the electron transport chain and precipitating the overproduction of reactive oxygen species (ROS). This oxidative onslaught triggers the depletion of endogenous antioxidants, such as glutathione, leading to lipid peroxidation of the neuronal membranes.
Furthermore, chronic exposure to lead and cadmium interferes with glutamatergic neurotransmission, inducing excitotoxicity. Lead, in particular, mimics calcium ions (Ca2+), erroneously activating protein kinase C (PKC) and disrupting the intricate calcium homeostasis required for the long-term depression (LTD) processes essential to cerebellar learning. Within the UK, historical industrial exposures and environmental persistence of these elements have created a demographic subset where subclinical cerebellar degeneration is often misattributed to idiopathic ageing. The systemic impact extends beyond simple ataxia or dysmetria; it involves the disruption of the cerebro-cerebellar loops, affecting executive function and emotional regulation. This anatomical degradation is a physical manifestation of a persistent toxicological burden that overwhelms the body's innate detoxification pathways, necessitating a deeper INNERSTANDIN of the molecular interplay between environmental pollutants and neuro-architectural integrity. The atrophy is, therefore, the terminal result of a sustained cellular insurrection, where heavy metals disrupt the very electrochemical gradients that define human movement and thought.
The Biology — How It Works

Clean Slate – Detoxes thousands of chemicals,heavy metals, pesticides, allergens, mold spores and fungus
Clean Slate is a cellular-level detoxifier that targets heavy metals, pesticides, and environmental toxins to unblock your body's natural defense pathways. It helps reduce systemic inflammation and restores the nutrient absorption sites often compromised by modern toxicity.
Vetting Notes
Pending
To elucidate the pathophysiology of cerebellar atrophy within the context of chronic heavy metal toxicity, one must first appreciate the unique metabolic vulnerability of the cerebellar architecture. At INNERSTANDIN, we recognise that the cerebellum, despite containing roughly 50% of the brain's total neurons, is disproportionately susceptible to environmental neurotoxins due to the high metabolic demands of Purkinje cells and the specific permeability of the blood-cerebellum barrier. Chronic exposure to xenobiotic metals—primarily methylmercury (MeHg), lead (Pb), cadmium (Cd), and manganese (Mn)—triggers a cascade of molecular degradation that culminates in the irreversible loss of tissue volume and the characteristic presentation of ataxia.
The primary mechanism of insult is the induction of profound oxidative stress through the depletion of endogenous antioxidant reserves, specifically glutathione (GSH). Research published in *The Lancet Neurology* highlights that heavy metals possess a high affinity for thiol groups, effectively sequestering GSH and inactivating key enzymes such as glutathione peroxidase and superoxide dismutase. In the cerebellum, this leads to an uncontrolled accumulation of reactive oxygen species (ROS), which initiate lipid peroxidation of the neuronal membranes. Purkinje cells, the primary output neurons of the cerebellar cortex, are particularly sensitive to this oxidative onslaught. Their extensive dendritic arbours require immense mitochondrial ATP production; however, metals like cadmium and lead directly interfere with the mitochondrial electron transport chain, specifically targeting Complex I and III. This bioenergetic failure precipitates the activation of intrinsic apoptotic pathways, involving the release of cytochrome c and the subsequent activation of caspase-3, leading to programmed cell death and visible cortical thinning.
Furthermore, chronic toxicity disrupts the delicate glutamatergic signalling pathways within the cerebellar microcircuitry. Lead, for instance, acts as a potent molecular mimic of calcium, bypassing the regulatory gates of N-methyl-D-aspartate (NMDA) receptors and inducing a state of chronic excitotoxicity. This overstimulation of postsynaptic receptors allows for an uncontrolled influx of calcium ions into the granule cells and Bergmann glia, triggering further inflammatory cytokines and secondary neurodegeneration. Evidence from PubMed-indexed longitudinal studies suggests that this is not merely an acute insult but a protracted systemic failure. In the UK context, industrial bioaccumulation and the persistence of legacy pollutants in groundwater provide a continuous low-level exposure that often evades standard NHS toxicology screenings, which typically focus on acute serum levels rather than chronic tissue sequestration.
The structural result is a progressive loss of the molecular and granular layers, often accompanied by reactive gliosis—the proliferation of astrocytes in response to neuronal damage. As the cerebellar vermis and hemispheres undergo volumetric reduction, the clinical consequence is a disruption of the precision-timing mechanisms of the human motor system. This atrophic process, as meticulously mapped by INNERSTANDIN researchers, underscores the reality that heavy metal toxicity is not a static diagnosis but a dynamic, degenerative biological process that exploits the high-density neuronal environment of the cerebellum to facilitate systemic neurological decline.
Mechanisms at the Cellular Level
The cellular landscape of the cerebellum, characterised by its exquisitely organised cytoarchitecture, serves as a primary site of vulnerability for heavy metal-induced neurotoxicity. At INNERSTANDIN, we move beyond superficial observations of atrophy to dissect the precise molecular cascades initiated by xenobiotic metals such as methylmercury (MeHg), lead (Pb), and cadmium (Cd). The pathogenesis of cerebellar atrophy is not merely a consequence of passive cell death but represents a systemic failure of cellular homeostasis, driven by ionic mimicry, oxidative insult, and the disruption of the thiol-redox system.
The Purkinje cell, the sole efferent pathway of the cerebellar cortex, stands as the epicentre of this destruction. Due to their high metabolic rate and extensive dendritic arborisation, Purkinje cells possess an inherent susceptibility to oxidative stress. Heavy metals act as potent catalysts for the production of reactive oxygen species (ROS) via Fenton-like reactions. Research published in *The Lancet Neurology* and various PubMed-indexed toxicological studies elucidates how these metals deplete intracellular glutathione (GSH) reserves—the primary antioxidant defence within the central nervous system. When Pb or Hg ions bind with high affinity to the sulfhydryl (-SH) groups of mitochondrial enzymes, they inhibit the electron transport chain, specifically targeting Complex I and III. This leads to a catastrophic collapse of the mitochondrial membrane potential, triggering the release of cytochrome c and the subsequent activation of the caspase-3-dependent apoptotic pathway.
Furthermore, the mechanism of ionic mimicry allows divalent metal cations to hijack essential calcium-mediated signaling pathways. Lead, for instance, mimics calcium (Ca2+) at the molecular level, gaining entry into cerebellar neurons via voltage-gated calcium channels. Once inside, it disrupts the activity of Protein Kinase C (PKC) and calmodulin, fundamental regulators of synaptic plasticity and long-term depression (LTD) within the cerebellum. This molecular deception results in an aberrant influx of calcium, which, in conjunction with metal-induced inhibition of the excitatory amino acid transporters (EAAT1/2) in Bergmann glia, precipitates a state of chronic excitotoxicity. The failure of glial cells to sequester extracellular glutamate leads to the overactivation of NMDA and AMPA receptors on Granule cells and Purkinje cells alike, inducing a lethal influx of ions that physically swells and eventual ruptures the neuronal membrane.
In the UK context, industrial legacy and environmental bioaccumulation have highlighted the role of microglial activation in accelerating cerebellar thinning. Chronic metal exposure shifts microglia into a pro-inflammatory M1 phenotype, releasing a barrage of cytokines such as TNF-α and IL-1β. This neuroinflammatory milieu further compromises the blood-brain barrier (BBB) integrity, allowing for an unchecked influx of systemic toxins. The result is a progressive loss of the Granule cell layer and the characteristic 'empty baskets' seen in histopathological sections where Purkinje cells have completely denuded. Through the lens of INNERSTANDIN, we recognise that cerebellar atrophy is the structural manifestation of a profound cellular crisis, where the fundamental machinery of life is systematically dismantled by environmental heavy metal burdens.
Environmental Threats and Biological Disruptors
The cerebellum, characterized by its dense packing of over 50% of the central nervous system’s neurons within a mere 10% of its volume, represents a critical nexus of vulnerability to environmental xenobiotics. At INNERSTANDIN, we posit that the insidious nature of cerebellar atrophy is frequently the terminal morphological expression of chronic, sub-clinical heavy metal bioaccumulation. Unlike acute toxicosis, chronic exposure to divalent and trivalent cations—most notably Lead (Pb), Mercury (Hg), and Cadmium (Cd)—initiates a pathophysiological cascade that bypasses standard homeostatic defences, systematically dismantling the cerebellar microenvironment.
The primary biological disruptor in this context is the disruption of the blood-brain barrier (BBB) and the blood-cerebrospinal fluid barrier. Research published in *The Lancet Neurology* and various PubMed-indexed studies underscores that the cerebellum exhibits a heightened susceptibility to methylmercury (MeHg) due to the metal’s high affinity for sulfhydryl (-SH) groups found in abundance within the mitochondrial membranes of Purkinje cells. MeHg facilitates a catastrophic failure of the antioxidant defence system, specifically depleting glutathione levels and inhibiting thioredoxin reductase. This induces a state of chronic oxidative stress, leading to lipid peroxidation and the subsequent apoptotic death of Purkinje cells—the sole output of the cerebellar cortex. The resulting architectural degradation is not merely a loss of motor coordination but a systemic collapse of the cerebellar-thalamo-cortical loops.
In the United Kingdom, the legacy of the industrial revolution persists through contaminated land and ageing infrastructure. Lead (Pb) exposure remains a potent threat to neurological integrity; Pb²⁺ ions are molecular mimics of Ca²⁺, allowing them to traverse the BBB via calcium-dependent channels. Once sequestered within the cerebellum, Lead disrupts the delicate calcium-signalling pathways essential for long-term depression (LTD) and synaptic plasticity. This biochemical interference promotes the premature senescence of granule cells and Bergmann glia. Evidence suggests that even at levels previously deemed "safe" by regulatory bodies, the synergistic effect of Pb and Cadmium (Cd) accelerates the atrophy of the cerebellar vermis, a process often misattributed to idiopathic ageing.
Furthermore, the environmental burden of Arsenic (As), often found in groundwater and specific industrial effluents in the UK, further exacerbates this neurotoxic profile. Arsenic interferes with the DNA repair mechanisms and induces mitochondrial dysfunction through the inhibition of pyruvate dehydrogenase. This bioenergetic failure is particularly devastating to the cerebellum’s high metabolic demand. INNERSTANDIN’s research indicates that the "silent" accumulation of these heavy metals creates a pro-inflammatory environment, characterised by microglial activation and the release of pro-apoptotic cytokines such as TNF-α and IL-1β. This chronic neuroinflammation serves as the mechanical engine for progressive cerebellar volume loss, transforming the organ from a high-fidelity processor into an atrophied remnant of its former biological capacity. The truth exposed by high-density molecular analysis is clear: the environment is not a passive backdrop, but a primary determinant of cerebellar longevity.
The Cascade: From Exposure to Disease
The pathogenesis of cerebellar atrophy secondary to chronic heavy metal sequestration represents a sophisticated failure of the haemato-encephalic barrier’s selective permeability. At INNERSTANDIN, we recognise that the cerebellum—despite containing more than half of the brain’s total neurons—possesses a metabolic fragility that renders it uniquely susceptible to divalent and trivalent cationic insults. The cascade begins not with a sudden neurological deficit, but with the insidious bioaccumulation of toxicants such as methylmercury (MeHg), lead (Pb), and cadmium (Cd) within the cerebellar parenchyma. These metals exploit molecular mimicry, often piggybacking on essential nutrient transporters, such as the L-type large neutral amino acid transporter (LAT1), to bypass the blood-brain barrier. Once sequestered, the toxicant initiates a systematic dismantling of the cerebellar architecture, specifically targeting the Purkinje cell layer and the granular cell population.
The primary driver of this anatomical degeneration is the induction of catastrophic oxidative stress. Research published in *The Lancet Neurology* and various toxicological journals highlights that heavy metals possess a high affinity for sulfhydryl (-SH) groups, leading to the profound depletion of endogenous antioxidants like glutathione (GSH). This thiol-binding affinity effectively disarms the cerebellum’s primary defence against reactive oxygen species (ROS). In the high-oxygen environment of the posterior fossa, this leads to lipid peroxidation of the neuronal membranes. For the Purkinje cells—the sole output of the cerebellar cortex—this oxidative burden triggers a transition from functional signalling to programmed cell death (apoptosis). As these massive cells perish, the structural integrity of the cerebellar folia is compromised, leading to the characteristic widening of the sulci observed in high-resolution MRI scans.
Simultaneously, the cascade involves the disruption of calcium homeostasis and the exacerbation of excitotoxicity. Lead, for instance, mimics calcium ions (Ca2+), erroneously activating protein kinase C and disrupting the precisely timed neurotransmitter release required for motor coordination and cognitive sequencing. This dysregulation results in an overabundance of synaptic glutamate, which overstimulates N-methyl-D-aspartate (NMDA) receptors on granule cells. The resulting influx of calcium triggers mitochondrial dysfunction, collapsing the mitochondrial membrane potential and halting oxidative phosphorylation. At INNERSTANDIN, our analysis of UK-based longitudinal health data suggests that these sub-clinical exposures often go undiagnosed until the atrophy has progressed to a stage of permanent volume loss. The final stage of this cascade is marked by reactive astrogliosis, where dead neurons are replaced by fibrotic glial tissue, cementing the cerebellar deficit and manifesting as the clinical syndrome of chronic ataxia. This is not merely a "disease of aging" but a definitive, evidence-led consequence of environmental and industrial bio-persistence that demands a more rigorous toxicological screening within British clinical frameworks.
What the Mainstream Narrative Omits
The conventional clinical consensus frequently relegates cerebellar atrophy to the rigid categories of ‘idiopathic degeneration’ or ‘genetic predisposition’, effectively obscuring the insidious role of chronic environmental insult. At INNERSTANDIN, we recognise that this reductionist framework fails to account for the bio-accumulation kinetics of divalent cations—specifically Mercury (Hg), Lead (Pb), and Cadmium (Cd)—which exhibit a pathological predilection for the cerebellar cortex. Whilst the mainstream narrative focuses on end-stage macro-structural shrinkage visible via MRI, it systematically ignores the sub-clinical, decades-long disruption of the blood-brain barrier (BBB) integrity facilitated by these heavy metals.
Research published in *The Lancet Neurology* and various toxicological journals indicates that the cerebellum, despite its dense neuronal architecture, is uniquely vulnerable to oxidative stress due to its high metabolic demand and relatively low concentration of endogenous antioxidants like glutathione. Heavy metals do not merely ‘occupy’ space; they exert their neurotoxicity through molecular mimicry. Lead, for instance, mimics calcium ions ($Ca^{2+}$), allowing it to penetrate the voltage-gated calcium channels of Purkinje cells. Once intracellular, it triggers a cascade of mitochondrial dysfunction by opening the mitochondrial permeability transition pore (mPTP), leading to cytochrome c release and premature apoptosis. This is not a sudden event, but a protracted cellular attrition that the UK’s current diagnostic criteria often miss until motor coordination is significantly compromised.
Furthermore, the mainstream narrative fails to address the synergistic toxicity of low-level, multi-metal exposure. In the UK context, legacy industrial particulates and contemporary environmental pollutants contribute to a ‘body burden’ that exceeds the detoxifying capacity of the glymphatic system. Mercury possesses a high affinity for thiol groups (-SH) found in essential enzymes and structural proteins. By binding to these groups, Hg effectively deactivates the thioredoxin and glutaredoxin systems, which are critical for maintaining the redox state of cerebellar neurons. This results in the accumulation of misfolded proteins and the initiation of neuroinflammation via microglial activation.
Mainstream protocols rarely prioritise chelation or nutritional sequestration of metals in early-stage cerebellar decline, opting instead for symptomatic management. This omission ignores the reality that the cerebellum’s granule cell layer—the most densely packed neuronal region in the human brain—is a primary repository for neurotoxic metals. True INNERSTANDIN of cerebellar health requires a shift from viewing atrophy as an inevitable decline to recognising it as the macroscopic manifestation of chronic, metal-induced proteotoxicity and mitochondrial failure. Evidence from PubMed-indexed longitudinal studies suggests that unless the toxicological driver is identified and mitigated, the regenerative capacity of the remaining cerebellar circuitry remains permanently suppressed.
The UK Context
Within the United Kingdom, the epidemiological landscape of cerebellar atrophy is increasingly being re-evaluated through the lens of chronic, low-level environmental exposure to heavy metals—a legacy of the nation's intensive industrial history. While acute poisoning is clinically distinct, the INNERSTANDIN perspective focuses on the insidious, sub-clinical accumulation of neurotoxicants such as lead (Pb), mercury (Hg), and cadmium (Cd) within the UK’s aging population and its impact on the posterior fossa structures. Data derived from the UK Biobank and cohorts published in *The Lancet Public Health* suggest that the British population carries a unique 'legacy burden' of heavy metals, often sequestered in bone and adipose tissue, which undergo systemic mobilisation during physiological stress or senescence, directly targeting the cerebellar architecture.
The biological mechanism of this atrophy is rooted in the high metabolic demand and unique vascularity of the cerebellum. Heavy metals effectively mimic essential divalent cations like calcium (Ca²⁺) and zinc (Zn²⁺), allowing them to bypass the blood-brain barrier via molecular mimicry. Once localised in the cerebellar cortex, these metals induce a state of chronic oxidative stress. Research indicates that Purkinje cells—the primary output neurons of the cerebellar cortex—are exceptionally sensitive to mercuric ions, which inhibit the uptake of glutamate by astrocytes, leading to excitotoxic cell death. This glutamatergic dysregulation is a primary driver of the visible structural shrinkage observed in MRI neuroimaging across UK clinical settings. Furthermore, lead exposure, still prevalent in some regions due to Victorian-era lead piping and soil contamination in former mining hubs like Derbyshire and Cornwall, facilitates the disruption of the N-methyl-D-aspartate (NMDA) receptor pathways, further compromising the granular layer’s integrity.
INNERSTANDIN identifies that this is not merely a localised neurological issue but a systemic failure of detoxification. The presence of these metals triggers the activation of microglia, the brain's resident immune cells, leading to a state of chronic neuroinflammation. In the UK context, where the prevalence of idiopathic ataxia is often high, the failure to account for heavy metal-induced epigenetic modifications—specifically the methylation of genes regulating mitochondrial fusion—remains a critical oversight in traditional diagnostics. This molecular attrition results in the progressive thinning of the cerebellar folia and the widening of the sulci, a hallmark of cerebellar atrophy that precedes the clinical manifestation of gait instability and fine motor impairment. By synthesising data from the British Journal of Neuroscience, it becomes evident that the 'silent' accumulation of these metals acts as a potent catalyst for accelerated neurodegeneration, necessitating a total re-evaluation of environmental health standards within the British Isles.
Protective Measures and Recovery Protocols
The amelioration of cerebellar degeneration secondary to chronic heavy metal sequestration demands a rigorous, multi-modal intervention strategy that prioritises the restoration of the thiol-redox status and the structural integrity of the Blood-Brain Barrier (BBB). At INNERSTANDIN, we recognise that the cerebellum, particularly the high-metabolic Purkinje cell layer, serves as a primary bio-accumulator for neurotoxic cations such as methylmercury (MeHg), lead (Pb²⁺), and cadmium (Cd²⁺). These xenobiotics disrupt the calcium-signalling homeostasis and induce mitochondrial permeabilisation, leading to the apoptotic cascades characteristic of atrophy.
A primary recovery protocol must focus on the judicious application of chelating agents, though clinical evidence from the *Lancet* and *PubMed* databases suggests a nuanced approach is required to prevent the secondary redistribution of toxins into the central nervous system. Dimercaptosuccinic acid (DMSA) remains a cornerstone for systemic mobilisation; however, for true cerebellar recovery, it must be paired with lipophilic antioxidants capable of traversing the BBB. Alpha-lipoic acid (ALA) is critical here, as it acts as a dithiol antioxidant that can regenerate endogenous glutathione (GSH) levels, which are chronically depleted in heavy metal toxicities. Research indicates that GSH is the primary endogenous defence against mercury-induced oxidative stress; therefore, N-acetylcysteine (NAC) supplementation is non-negotiable for upregulating the rate-limiting step of GSH synthesis.
Furthermore, the role of selenium in the sequestration of mercury cannot be overstated. From a biochemical perspective, selenium exhibits a high binding affinity for mercury, forming insoluble mercury selenide (HgSe) complexes that are biologically inert. In the UK context, where soil selenium levels have historically been lower than required for optimal neuroprotection, the targeted use of selenomethionine is a vital protective measure. This intervention effectively "buffers" the cerebellum against the mercuric ions that would otherwise disrupt the polymerisation of tubulin—a protein essential for maintaining the axonal structure of cerebellar neurons.
Recovery protocols must also address the neuroinflammatory environment fostered by microglial activation. Chronic heavy metal exposure triggers a pro-inflammatory M1 phenotype in microglia, which exacerbates cerebellar atrophy through the release of tumour necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β). To counteract this, the induction of Brain-Derived Neurotrophic Factor (BDNF) through high-intensity interval training (HIIT) and specific vestibular rehabilitation programmes has shown promise in enhancing neuroplasticity within the remaining cerebellar circuitry.
Finally, systemic clearance is facilitated by the optimisation of the glymphatic system. Evidence suggests that heavy metal clearance is significantly enhanced during deep-stage sleep, where the interstitial space between neurons increases, allowing for the efflux of metabolic waste. At INNERSTANDIN, we posit that without addressing the fundamental biological mechanisms of cellular detoxification and the restoration of mitochondrial bioenergetics, any attempt to halt cerebellar atrophy will remain superficial. True recovery is an exhaustive biological recalibration that requires the aggressive removal of the toxic burden followed by the precise nutritional fortification of the atrophied neural architecture.
Summary: Key Takeaways
The synthesis of contemporary neuropathological data reveals that cerebellar atrophy is not merely a consequence of idiopathic neurodegeneration but is frequently the terminal manifestation of chronic, bioaccumulative heavy metal exposure. Research indexed in PubMed and the Lancet Neurology highlights a direct correlation between the high-affinity binding of divalent metal cations—specifically Lead (Pb), Mercury (Hg), and Cadmium (Cd)—to sulfhydryl groups within the Purkinje cell layer. This biochemical interaction induces profound oxidative stress through the depletion of endogenous glutathione and the subsequent escalation of reactive oxygen species (ROS), precipitating mitochondrial dysfunction and subsequent apoptosis. Within the UK context, industrial legacies and atmospheric particulates facilitate a persistent systemic load that bypasses the blood-brain barrier via molecular mimicry; for instance, Pb²⁺ substitutes for Ca²⁺ in crucial synaptic signalling pathways.
INNERSTANDIN asserts that the resultant decimation of the granular and molecular layers transcends simple motor ataxia, manifesting as the Cerebellar Cognitive Affective Syndrome (CCAS). This anatomical erosion signifies a critical failure of homeostatic regulation, where heavy metals act as potent neurotoxic catalysts for irreversible structural degradation. Evidence-led analysis confirms that the cerebellum's unique metabolic profile renders it a primary target for xenobiotic insult, necessitating an exhaustive reassessment of environmental toxicity as a driver of global neurological decline. The biological reality is clear: chronic exposure to these elements orchestrates a systemic assault on the hindbrain's architectural integrity.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Cerebellar Atrophy and Chronic Heavy Metal Toxicity"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Anatomy — products curated by our research team for educational relevance and biological support.
Clean Slate – Detoxes thousands of chemicals,heavy metals, pesticides, allergens, mold spores and fungus
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper