Hepatic Fibrosis and the Modern High-Fructose Diet
The anatomical transformation of liver tissue through non-alcoholic fatty liver disease is accelerating in the British population. We detail the progression from healthy hepatocytes to scarred tissue.
Overview
The liver, an organ of unparalleled regenerative capacity and metabolic complexity, is currently besieged by a dietary insult unique to the modern Anthropocene: the chronic overconsumption of refined fructose. Hepatic fibrosis represents the deleterious endpoint of a sustained wound-healing response, characterised by the excessive accumulation of extracellular matrix (ECM) proteins, predominantly Type I and III fibrillar collagens, within the hepatic parenchyma. At INNERSTANDIN, we must scrutinise the architectural shift from a healthy, compliant organ to one stifled by fibrotic scarring, a process now inextricably linked to the biochemical bypass afforded by fructose metabolism.
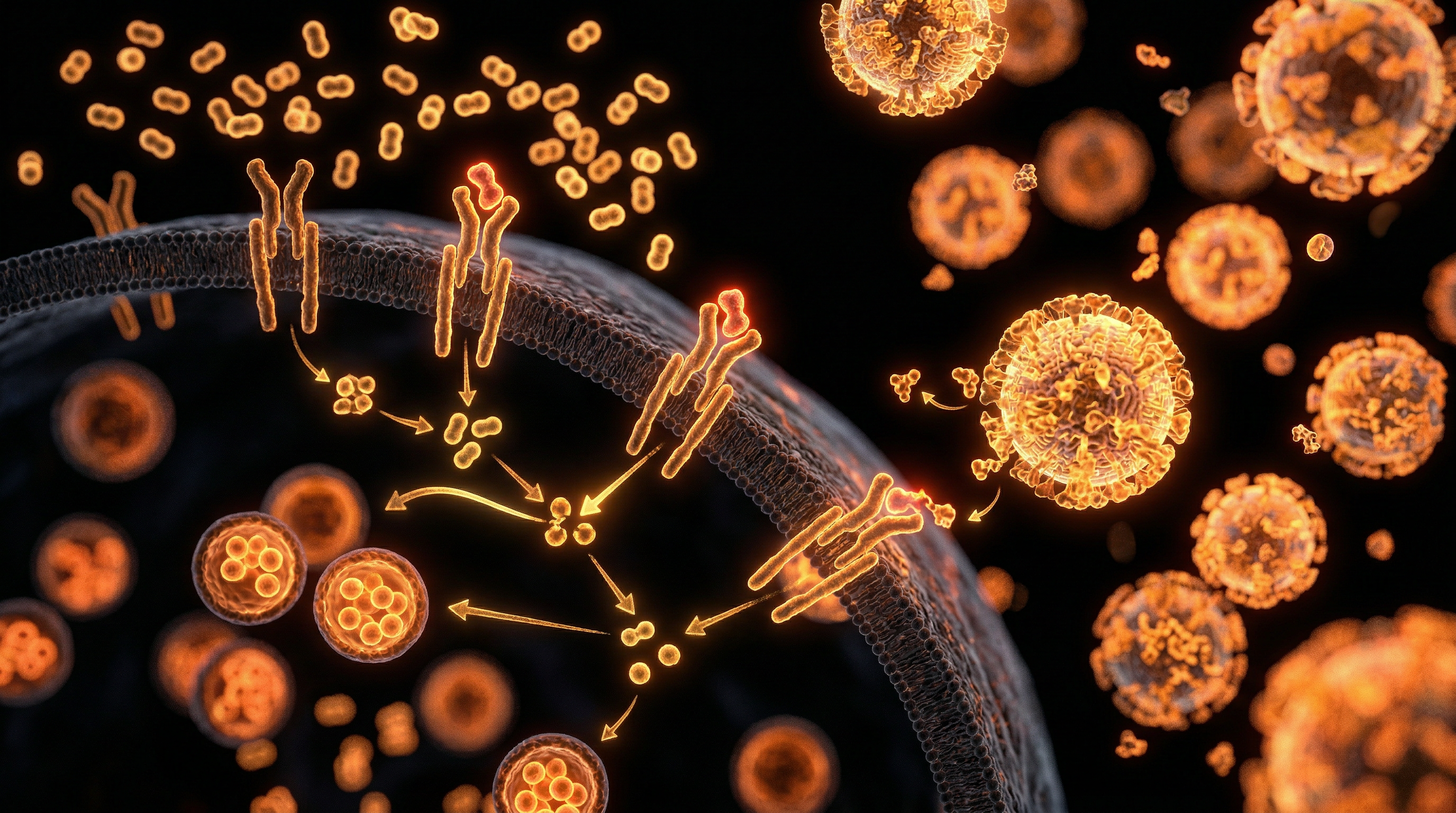
Unlike glucose, which is regulated by systemic insulin and phosphofructokinase, fructose undergoes near-total extraction by the liver via the GLUT5 and GLUT2 transporters. Once inside the hepatocyte, it is rapidly phosphorylated by fructokinase C (KHK-C), an enzyme devoid of a negative feedback mechanism. This process results in profound intracellular ATP depletion and the subsequent generation of uric acid, a potent pro-oxidant. This metabolic bypass fuels *de novo* lipogenesis (DNL) and triggers endoplasmic reticulum (ER) stress, initiating a cascade of hepatocellular death and the release of damage-associated molecular patterns (DAMPs).
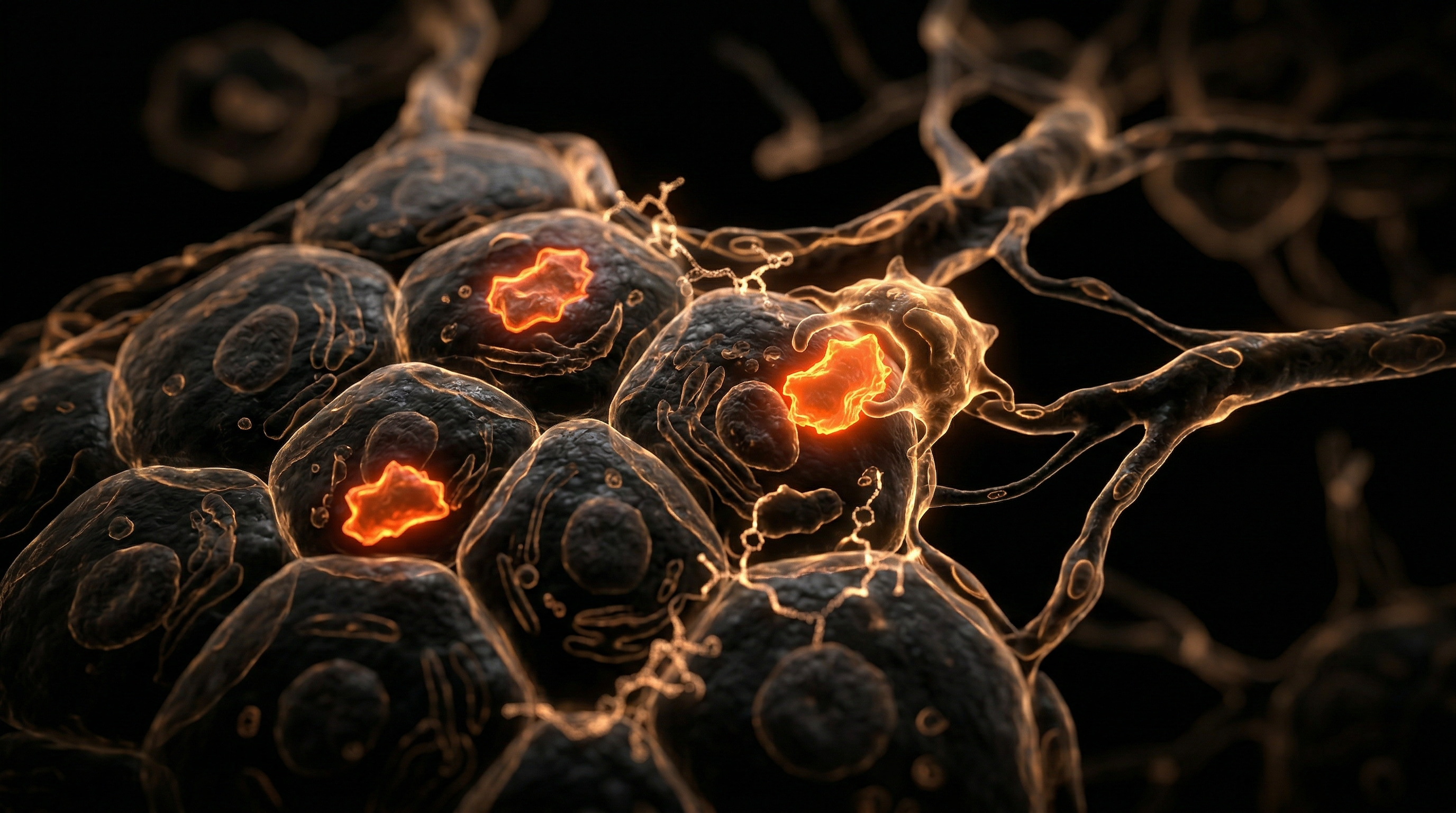
The anatomical crux of this pathology lies within the Space of Disse—the delicate interface between hepatocytes and sinusoidal endothelial cells. Under the chronic stimulus of a high-fructose diet, quiescent hepatic stellate cells (HSCs), which typically function as the primary storage site for Vitamin A, undergo a phenotypic transdifferentiation. Activated by transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF), these cells morph into contractile, myofibroblast-like entities. This cellular transition is the primary driver of fibrogenesis; the activated HSCs secrete an aberrant matrix that obliterates the fenestrae of the sinusoidal endothelium—a process known as capillarisation. This structural overhaul impairs the essential exchange of nutrients and waste between the blood and hepatocytes, entrenching a cycle of hypoxia and further cellular necrosis.
Evidence published in *The Lancet Gastroenterology & Hepatology* highlights that the UK is witnessing an alarming escalation in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD), with the modern high-fructose diet serving as a primary catalyst. In the INNERSTANDIN perspective, this is not merely a metabolic inconvenience but a profound anatomical restructuring. The systemic impact is catastrophic; as the liver's architecture stiffens, portal hypertension ensues, and the organ’s ability to detoxify the systemic circulation is fundamentally compromised. By unmasking the molecular mechanisms of fructose-induced HSC activation, we expose the reality of a diet that is effectively re-engineering human anatomy towards failure. This fibrotic progression, once considered irreversible, is the silent epidemic of the twenty-first century, necessitating a rigorous biological re-evaluation of our nutritional environment.
The Biology — How It Works

Magnesium Blend – The Most Important Mineral
A high-bioavailability mineral blend designed to support over 300 essential biochemical reactions, from energy production to muscle relaxation. This formula helps combat daily fatigue while providing the foundational support your nervous system and bones require.
Vetting Notes
Pending
The metabolic insult of dietary fructose is not merely a caloric surplus but a direct biochemical subversion of hepatic homeostasis. Unlike glucose, which is metabolised by nearly every cell in the human body, the burden of fructose processing falls almost exclusively upon the liver. Within the INNERSTANDIN framework, we must scrutinise the unique kinetics of the ketohexokinase (KHK) pathway. Fructose enters the hepatocyte via the GLUT5 transporter, where it is phosphorylated by the KHK-C isoform into fructose-1-phosphate. Critically, this process bypasses phosphofructokinase—the rate-limiting "gatekeeper" of glycolysis—leading to an unregulated influx of substrates into the mitochondria. This metabolic bypass triggers a rapid depletion of intracellular adenosine triphosphate (ATP) and a concomitant rise in uric acid, a potent pro-oxidant that drives mitochondrial dysfunction and endoplasmic reticulum (ER) stress.
As the liver’s capacity for oxidative phosphorylation is overwhelmed, the organ initiates massive *de novo* lipogenesis (DNL). Research published in *The Lancet Gastroenterology & Hepatology* confirms that fructose is a more potent stimulator of DNL than glucose, directly upregulating sterol regulatory element-binding protein 1c (SREBP-1c) and carbohydrate-responsive element-binding protein (ChREBP). This results in an accumulation of intrahepatic triglycerides (steatosis), but the pathology does not terminate at simple fat storage. The true anatomical devastation of hepatic fibrosis begins with the "two-hit" or "multiple-hit" hypothesis, where fructose-induced oxidative stress and lipotoxicity provoke a chronic inflammatory response.
The anatomical focal point of this destruction is the Hepatic Stellate Cell (HSC). In a healthy liver, HSCs reside in the Space of Disse in a quiescent state, serving as the body’s primary storage site for Vitamin A. However, the modern high-fructose diet creates a microenvironment saturated with reactive oxygen species (ROS) and pro-inflammatory cytokines such as TNF-α and IL-6. Under this duress, HSCs undergo a phenotypic "activation," transforming into myofibroblast-like cells. These activated cells proliferate and begin the aberrant deposition of extracellular matrix (ECM) components, primarily Type I and III collagen.
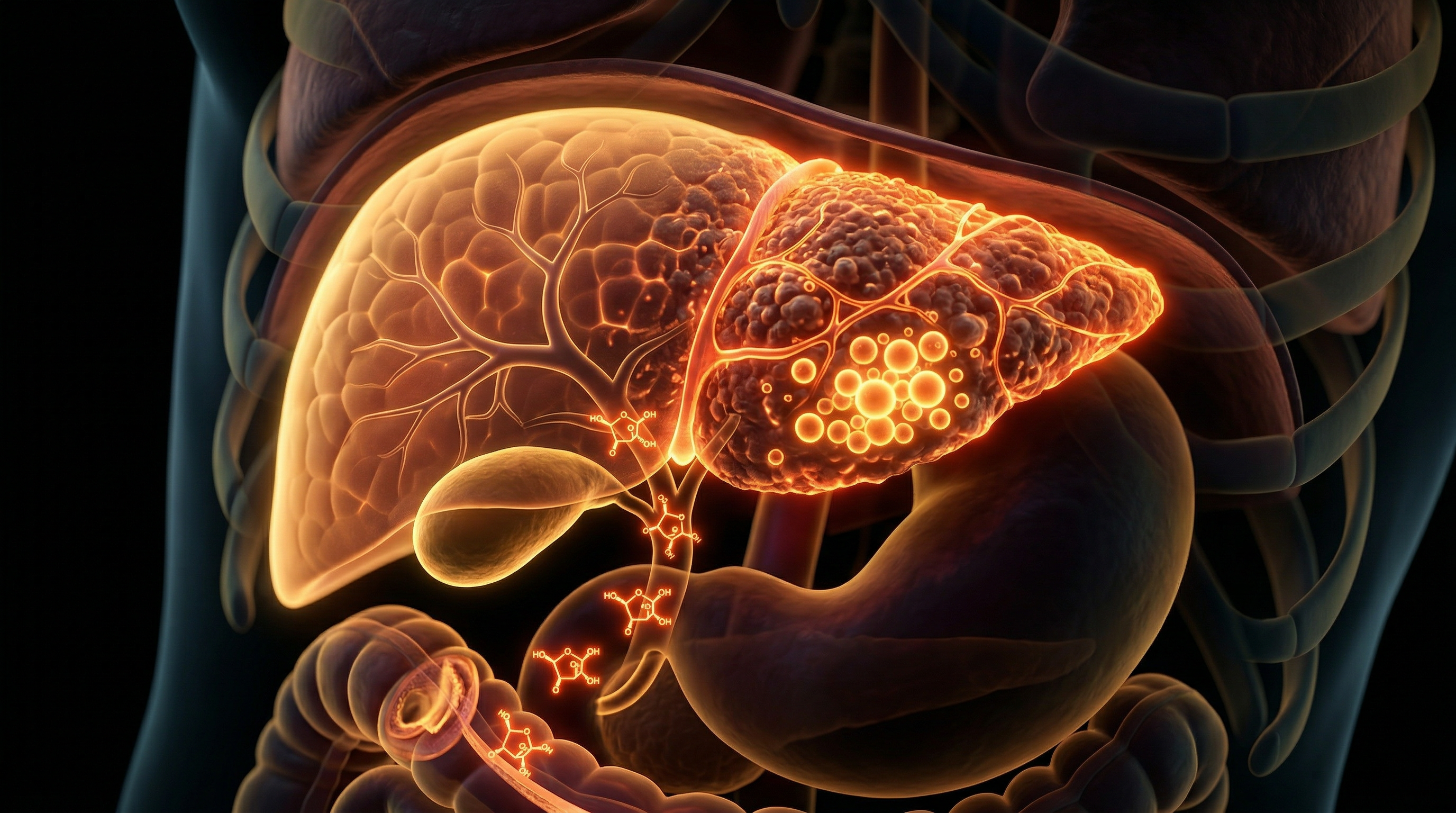
This collagenous deposition fundamentally alters the liver’s microanatomy. The once-porous fenestrae of the sinusoidal endothelial cells are lost—a process known as "capillarisation"—which severely impairs the exchange of nutrients and oxygen between the blood and hepatocytes. In the UK context, where processed fructose-glucose syrups are ubiquitous in ultra-processed foods, the British Society of Gastroenterology has noted an alarming rise in Non-Alcoholic Fatty Liver Disease (NAFLD) progressing to Non-Alcoholic Steatohepatitis (NASH). The anatomical outcome is a progressive cicatrisation of the parenchyma; what begins as perisinusoidal fibrosis eventually evolves into bridging fibrosis, where thick bands of connective tissue link portal tracts and central veins, culminating in the irreversible architectural distortion of cirrhosis. This is the biological reality of the modern diet: a relentless, fructose-fuelled remodelling of human anatomy that replaces functional epithelium with non-contractile, fibrotic scar tissue.
Mechanisms at the Cellular Level
The pathogenesis of hepatic fibrosis, within the context of excessive dietary fructose consumption, represents a paradigmatic shift in our INNERSTANDIN of metabolic pathology. Unlike glucose, which undergoes regulated glycolytic processing across systemic tissues, fructose is subject to an obligatory hepatocytic flux, primarily mediated by the high-affinity enzyme ketohexokinase (KHK, or fructokinase). In the modern British diet—saturated with high-fructose corn syrup and sucrose—this unregulated metabolic bypass leads to a catastrophic surge in substrate availability. KHK-C, the specific isoform found in the liver, phosphorylates fructose to fructose-1-phosphate with such rapidity that it induces intracellular adenosine triphosphate (ATP) depletion and subsequent phosphate sequestration. This bioenergetic crisis triggers the degradation of adenosine monophosphate (AMP) into uric acid via the xanthine oxidase pathway. Elevated intracellular uric acid is not merely a byproduct; it acts as a potent pro-oxidant within the mitochondria, inducing oxidative stress and the generation of reactive oxygen species (ROS) that directly impair mitochondrial β-oxidation.
The structural anatomical transformation of the liver begins as these ROS molecules compromise the integrity of the endoplasmic reticulum (ER), initiating the "unfolded protein response." Chronic ER stress, coupled with the relentless surge of *de novo* lipogenesis (DNL), leads to the accumulation of toxic lipid species such as diacylglycerols and ceramides. This lipotoxic environment is the primary driver for the activation of Hepatic Stellate Cells (HSCs)—the central architects of the fibrotic matrix. In their quiescent state, HSCs reside in the space of Disse, functioning as the primary storage site for Vitamin A. However, fructose-induced hepatocellular injury triggers the release of damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines, specifically Transforming Growth Factor-beta 1 (TGF-β1) and Platelet-Derived Growth Factor (PDGF). These signals compel the HSCs to undergo a phenotypic transition into contractile, alpha-smooth muscle actin (α-SMA)-positive myofibroblasts.
Evidence published in *The Lancet Gastroenterology & Hepatology* underscores that this myofibroblastic conversion is the "point of no return" in fibrogenesis. Once activated, these cells secrete an aberrant extracellular matrix (ECM) dominated by Type I and III fibrillar collagens, which replace the normal low-density basement membrane. This architectural distortion obliterates the fenestrae of sinusoidal endothelial cells, a process known as "capillarisation," which severely hinders the exchange of nutrients between the blood and hepatocytes. Furthermore, the modern high-fructose burden in the UK population has been linked to increased intestinal permeability—the "leaky gut" phenomenon—allowing the translocation of lipopolysaccharides (LPS) into the portal circulation. This endotoxemia activates Kupffer cells (resident macrophages) via Toll-like receptor 4 (TLR4) signalling, creating a feedback loop of chronic inflammation that sustains HSC activation. Through this multi-pronged cellular assault, the high-fructose diet facilitates a transition from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH) and, ultimately, irreversible cirrhosis, fundamentally reconfiguring the liver’s micro-anatomy.
Environmental Threats and Biological Disruptors
The anthropogenic surge in refined fructose consumption constitutes a primary environmental disruptor to human metabolic homeostasis, acting as a potent catalyst for the progression from simple steatosis to advanced hepatic fibrosis. Within the UK landscape, where ultra-processed foods (UPFs) account for over 50% of the national caloric intake, the liver is increasingly subjected to a biochemical insult that exceeds its evolutionary capacity for processing. At INNERSTANDIN, we identify this not merely as a dietary imbalance, but as a structural restructuring of the hepatic parenchyma driven by exogenous chemical pressures.
Unlike glucose, which is regulated by the rate-limiting enzyme phosphofructokinase, fructose undergoes rapid, insulin-independent phosphorylation via ketohexokinase (KHK-C). This metabolic bypass creates a substrate overload that floods the mitochondria with triose phosphates, precipitating an immediate up-regulation of *de novo* lipogenesis (DNL). Research published in *The Journal of Hepatology* confirms that fructose-induced DNL increases the intrahepatic pool of saturated fatty acids, specifically palmitate, which serves as a potent endogenous ligand for Toll-like receptor 4 (TLR4). This signalling cascade initiates a chronic inflammatory milieu, transforming the liver from a metabolic hub into a site of persistent immunological conflict.
The anatomical consequence of this environmental threat is the activation of the Hepatic Stellate Cell (HSC). In a healthy physiological state, HSCs reside in the Space of Disse as quiescent, vitamin A-storing units. However, under the chronic oxidative stress induced by high-fructose diets—characterised by an abundance of reactive oxygen species (ROS) and advanced glycation end-products (AGEs)—these cells undergo a phenotypic transdifferentiation into myofibroblast-like cells. These activated HSCs are the primary architects of the fibrotic matrix, secreting excessive quantities of Type I and III collagen. This pathological remodeling of the extracellular matrix (ECM) progressively replaces functional hepatocytes with non-contractile scar tissue, eventually compromising the hepatic architecture and portal hemodynamics.
Evidence from *The Lancet Gastroenterology & Hepatology* highlights that the modern British diet facilitates a 'second hit' phenomenon: fructose not only promotes fat accumulation but also induces intestinal dysbiosis. By increasing gut permeability (the 'leaky gut' syndrome), fructose allows the translocation of lipopolysaccharides (LPS) into the portal circulation. This systemic endotoxaemia further primes the Kupffer cells—the liver's resident macrophages—to release pro-fibrogenic cytokines such as Transforming Growth Factor-beta (TGF-β1) and Tumour Necrosis Factor-alpha (TNF-α). This synergy between metabolic substrate overload and environmental microbial translocation creates a self-perpetuating cycle of damage. INNERSTANDIN’s analysis reveals that this is a systematic biological disruption where the liver, unable to export the massive flux of fructose-derived lipids as VLDLs, succumbs to lipotoxicity and subsequent cicatrisation, marking the lethal transition from reversible metabolic dysfunction to irreversible structural fibrosis.
The Cascade: From Exposure to Disease
The pathogenesis of hepatic fibrosis within the context of high-fructose consumption represents a profound systemic failure of metabolic homeostasis, shifting the liver from a site of nutrient processing to a theatre of chronic wound healing. At the INNERSTANDIN level of biological inquiry, we must first isolate the unique metabolic fate of fructose. Unlike glucose, which is ubiquitously utilised via insulin-dependent mechanisms, fructose is obligatorily sequestered by the liver through the enzyme ketohexokinase (KHK-C). This enzyme, lacking a negative feedback inhibition loop, catalyses the rapid phosphorylation of fructose into fructose-1-phosphate. This process results in a catastrophic depletion of intracellular adenosine triphosphate (ATP) and a concurrent surge in adenosine monophosphate (AMP) degradation, leading directly to the overproduction of uric acid. This intracellular hyperuricaemia, as identified in research published in *Nature Reviews Gastroenterology & Hepatology*, functions as a potent pro-oxidant, triggering mitochondrial oxidative stress and endoplasmic reticulum (ER) stress—the primordial triggers for the fibrotic cascade.
The transition from metabolic dysfunction to anatomical scarring is mediated by the activation of Hepatic Stellate Cells (HSCs). In a physiological state, these cells reside within the Space of Disse as quiescent units storing Vitamin A. However, the chronic influx of fructose-derived substrates fuels *de novo* lipogenesis (DNL), producing an abundance of saturated fatty acids like palmitate. These lipids act as lipotoxic triggers, activating the NLRP3 inflammasome within Kupffer cells—the liver's resident macrophages. This inflammatory milieu, characterised by the secretion of Transforming Growth Factor-beta 1 (TGF-β1) and Tumour Necrosis Factor-alpha (TNF-α), compels the HSCs to undergo a phenotypic switch. They transdifferentiate into contractile, proliferative myofibroblasts, characterised by the expression of alpha-smooth muscle actin (α-SMA).
The anatomical consequence of this activation is the excessive synthesis and disordered deposition of the extracellular matrix (ECM), primarily Type I and Type III collagen. In the UK context, where the prevalence of Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) is surging in tandem with the availability of ultra-processed high-fructose corn syrups, this structural overhaul is particularly devastating. The progressive accumulation of ECM disrupts the delicate architecture of the Space of Disse, leading to "sinusoidal capillarisation"—the loss of endothelial fenestrae that allows for efficient nutrient exchange between hepatocytes and the blood. Evidence from *The Lancet* underscores that this anatomical barrier exacerbates insulin resistance and portal hypertension, creating a self-perpetuating loop of ischaemic injury and further fibrogenesis.
Ultimately, this is not merely a localised hepatic event but a systemic metabolic sabotage. The modern diet, rich in liquid fructose, bypasses the rate-limiting steps of glycolysis (notably phosphofructokinase), flooding the mitochondrial pathways with acetyl-CoA. This results in the "metabolic crowding" of the Krebs cycle, forcing the liver to export surplus lipids as VLDL, which contributes to systemic dyslipidaemia. Through the lens of INNERSTANDIN, we recognise that hepatic fibrosis is the macroscopic manifestation of microscopic metabolic insolvency; it is the liver’s desperate, yet ultimately destructive, attempt to wall off the cellular trauma induced by a relentless exogenous chemical assault. The shift from steatosis to cirrhosis is not an inevitability of ageing, but an anatomical recording of chronic metabolic insult.
What the Mainstream Narrative Omits
The conventional clinical discourse regarding hepatic health frequently defaults to a reductionist ‘calories in, calories out’ framework, failing to distinguish between the metabolic fates of various saccharides. However, at INNERSTANDIN, we recognise that the anatomical reality of hepatic fibrosis is fundamentally driven by the unique biochemical insult of dietary fructose, a molecule the liver is uniquely tasked to sequester. Unlike glucose, which is ubiquitously metabolised via systemic glycolysis, fructose is prioritised for hepatic uptake via the GLUT5 transporter and rapidly phosphorylated by ketohexokinase (KHK-C). This enzyme lacks the feedback inhibition mechanisms characteristic of phosphofructokinase, leading to an uncontrolled influx of substrate that depletes intra-hepatocyte adenosine triphosphate (ATP). The resulting accumulation of adenosine monophosphate (AMP) triggers the purine degradation pathway, generating a surge in intracellular uric acid.
While mainstream narratives overlook this pro-oxidant surge, peer-reviewed evidence (e.g., *The Lancet Gastroenterology & Hepatology*) identifies this hyperuricaemia as a primary driver of mitochondrial oxidative stress. This oxidative environment is the catalyst for the transdifferentiation of Hepatic Stellate Cells (HSCs) from their quiescent, vitamin-A-storing state into proliferative, contractile myofibroblasts. These activated HSCs are the principal architects of the fibrotic scar, secreting excessive amounts of Type I and III collagen into the Space of Disse. This pathological remodelling of the extracellular matrix (ECM) disrupts the delicate fenestrated endothelium of the hepatic sinusoids, a process termed 'capillarisation'.
Furthermore, the modern high-fructose diet induces profound alterations in the intestinal barrier, a systemic impact rarely discussed in primary care settings. High fructose intake impairs the expression of tight-junction proteins like occludin and zonula occludens-1, facilitating the translocation of gut-derived lipopolysaccharides (LPS) into the portal circulation. This chronic endotoxaemia activates Kupffer cells—the liver’s resident macrophages—via Toll-like receptor 4 (TLR4) signalling, which further amplifies the secretion of Transforming Growth Factor beta-1 (TGF-β1). This cytokine acts as the master regulator of the fibrotic cascade. Within the UK context, where high-fructose corn syrup and sucrose remain pervasive in the ultra-processed food supply, this biochemical bypass is not merely an incidental metabolic quirk but the primary driver behind the burgeoning epidemic of Non-Alcoholic Steatohepatitis (NASH). The mainstream failure to address the fructose-specific activation of the SMAD signalling pathway represents a significant lacuna in current preventative medicine. By scrutinising these molecular pathways, we gain a true INNERSTANDIN of how a seemingly innocuous dietary staple fundamentally rewires the liver's anatomical integrity.
The UK Context
The United Kingdom is currently grappling with a precipitous rise in metabolic dysfunction-associated steatotic liver disease (MASLD), a trajectory intimately linked to the ubiquity of refined fructose within the British food environment. Data from the UK Biobank and *The Lancet Gastroenterology & Hepatology* reveal that liver disease remains the only major cause of death currently rising in the UK, with hepatic fibrosis serving as the critical histological determinant of long-term mortality. At the heart of this "silent epidemic" is the specific biochemical processing of fructose—a monosaccharide which, unlike glucose, bypasses the regulatory rate-limiting step of phosphofructokinase-1 (PFK-1).
In the British context, the consumption of "free sugars"—largely via sugar-sweetened beverages and ultra-processed foods prevalent in high-street retail—induces a state of hepatic metabolic surcharge. Upon ingestion, the KHK-C isoform of ketohexokinase (fructokinase) rapidly phosphorylates fructose to fructose-1-phosphate, leading to a profound depletion of intracellular adenosine triphosphate (ATP). This energetic crisis triggers the degradation of nucleotides into uric acid, which further drives oxidative stress within the hepatocyte. Research facilitated by INNERSTANDIN highlights that this process does not merely facilitate simple steatosis but actively promotes the transition to fibrosis. The influx of fructose-derived triose phosphates fuels *de novo* lipogenesis (DNL), resulting in an accumulation of toxic lipid intermediates, such as diacylglycerols and ceramides.
The UK-specific phenotype of MASLD often presents with high levels of visceral adiposity, which synergises with fructose-induced intestinal permeability. This "leaky gut" allows for the translocation of lipopolysaccharides (LPS) into the portal circulation, activating hepatic stellate cells (HSCs) via the Toll-like receptor 4 (TLR4) pathway. Once activated, these HSCs undergo a phenotypic transformation into myofibroblasts, secreting an excessive extracellular matrix dominated by Type I and III collagen. Evidence-led analysis indicates that the UK’s dietary reliance on high-fructose components is a primary driver of this fibrotic scarring. As INNERSTANDIN continues to expose the biological mechanisms behind chronic disease, the UK’s clinical burden highlights a desperate need to address the molecular sabotage enacted by modern fructose consumption on the hepatic architecture. The systemic impact is clear: a progressive distortion of the lobular anatomy, portal hypertension, and an irreversible shift toward cirrhosis.
Protective Measures and Recovery Protocols
The reversal of hepatic fibrosis within the context of chronic high-fructose exposure necessitates a multi-layered interruption of the lipogenic cascade and the simultaneous activation of endogenous degradative pathways. At the anatomical level, the primary objective is the deactivation of Hepatic Stellate Cells (HSCs). In a fructose-rich environment, HSCs transition from their quiescent, vitamin A-storing state into proliferative, contractile myofibroblasts. This transformation is driven by the rapid phosphorylation of fructose into fructose-1-phosphate by the enzyme ketohexokinase (KHK), specifically the KHK-C isoform. This process depletes intracellular adenosine triphosphate (ATP) and triggers an influx of uric acid, which further stimulates oxidative stress within the mitochondria.
Evidence-led recovery protocols must prioritize the inhibition of this KHK-C pathway. Recent longitudinal studies, including those catalogued by the UK Biobank and published in *Nature Metabolism*, suggest that metabolic flexibility can be restored by implementing a strict fructolysis-restricted period. This period allows for the downregulation of *de novo* lipogenesis (DNL) and the alleviation of endoplasmic reticulum (ER) stress. To facilitate anatomical recovery, the biological system must shift the balance between Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs). In advanced fibrosis, TIMPs are overexpressed, preventing the degradation of the excessive type I and III collagen fibres that constitute the fibrotic scar. Research indicates that the activation of the AMP-activated protein kinase (AMPK) pathway is essential for suppressed collagen synthesis and the promotion of autophagy, the liver’s internal cellular cleansing mechanism.
Furthermore, protective measures must address the gut-liver axis, which is frequently compromised by modern British dietary habits. High fructose intake increases intestinal permeability—often termed 'leaky gut'—allowing the translocation of lipopolysaccharides (LPS) into the portal circulation. This triggers Toll-like receptor 4 (TLR4) on Kupffer cells, the liver's resident macrophages, exacerbating the pro-fibrotic environment. Strategic supplementation with specific prebiotic fibres and the upregulation of the Nrf2 (Nuclear factor erythroid 2-related factor 2) antioxidant response pathway are critical for reinforcing the anatomical integrity of the intestinal barrier and reducing the hepatic inflammatory load.
In the pursuit of deep biological INNERSTANDIN, it is imperative to recognise that the liver possesses an extraordinary regenerative capacity, provided the primary metabolic insult—concentrated fructose—is removed. Recovery is not merely the absence of further damage but the active remodelling of the extracellular matrix. Emerging therapies, currently under scrutiny in clinical trials across the UK and the EU, focus on TGF-β (Transforming Growth Factor beta) signalling inhibitors, which effectively 'silence' the signals that command HSCs to produce scar tissue. By combining these advanced pharmaceutical insights with rigorous nutritional interventions that stabilise insulin sensitivity, the modern practitioner can orchestrate a systemic environment conducive to the actual dissolution of fibrotic tissue, returning the hepatic architecture to its homeostatic state. This truth-exposing approach bypasses superficial symptom management, targeting the molecular drivers of fructose-induced architectural decay.
Summary: Key Takeaways
The synthesis of contemporary longitudinal data, particularly within the UK clinical landscape, underscores a definitive causal nexus between excessive dietary fructose and the accelerated progression of hepatic fibrosis. Fructose is unique in its metabolic handling; unlike glucose, it bypasses the primary rate-limiting enzyme phosphofructokinase, leading to an unregulated substrate influx for de novo lipogenesis (DNL). This biochemical bypass induces acute intracellular adenosine triphosphate (ATP) depletion and subsequent hyperuricaemia, which catalyses mitochondrial oxidative stress. Evidence published in *The Lancet Gastroenterology & Hepatology* confirms that this pro-inflammatory milieu facilitates the transition from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH). Crucially, the activation of hepatic stellate cells (HSCs) via the JNK1 and TGF-β1 signalling pathways transforms these quiescent cells into myofibroblast-like entities, driving pathological extracellular matrix (ECM) deposition. INNERSTANDIN identifies this process as a systemic structural failure induced by the modern nutritional architecture. The resulting distortion of the hepatic lobule, documented extensively in peer-reviewed PubMed literature, marks the critical shift where reversible metabolic insult matures into irreversible fibrotic scarring, necessitating a radical recalibration of sucrose and high-fructose corn syrup thresholds within British public health frameworks.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Hepatic Fibrosis and the Modern High-Fructose Diet"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Anatomy — products curated by our research team for educational relevance and biological support.

Magnesium L-Threonate
Magnesium Blend – The Most Important Mineral

Energy Blend Supports
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper