Iron Anemia: The Hepcidin Regulatory Paradox
Standard iron supplementation often fails due to the hepcidin block caused by systemic inflammation and poor gut health. This report clarifies the difference between iron intake and iron utilization in the UK population.
Overview
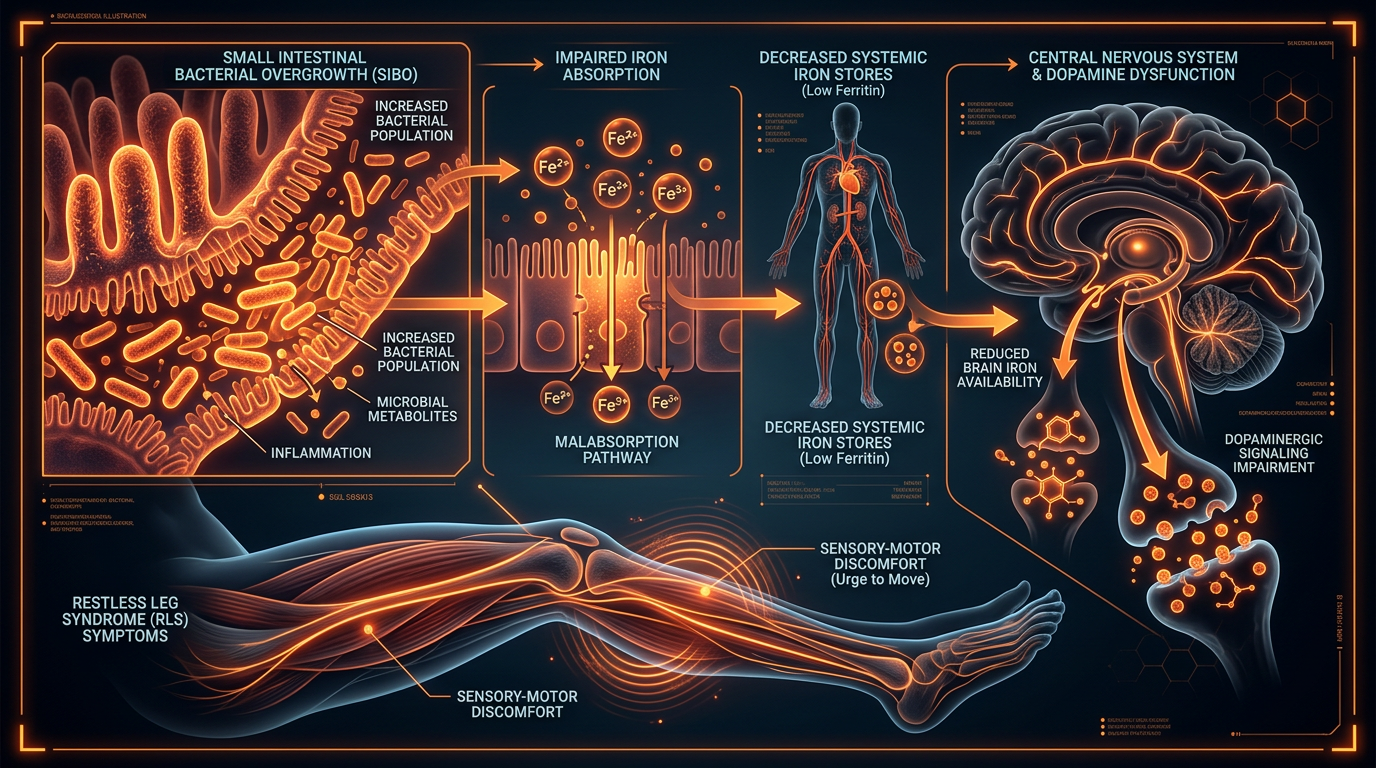
The clinical landscape of iron deficiency has long been dominated by a reductionist paradigm, framing anaemia as a simple deficit in dietary intake or a consequence of chronic haemorrhagic loss. However, through the lens of INNERSTANDIN, we must confront a far more complex biological reality: the Hepcidin Regulatory Paradox. This phenomenon represents a systemic failure of iron trafficking, where the physiological requirement for iron is subverted by a molecular "iron-lock" mechanism, rendering even substantial iron stores biologically unavailable for erythropoiesis. At the centre of this paradox is hepcidin, a 25-amino acid peptide hormone synthesised primarily by hepatocytes, which serves as the master regulator of systemic iron homeostasis.
Hepcidin functions by binding to ferroportin—the sole known mammalian cellular iron exporter found on the basolateral membrane of duodenal enterocytes and the plasma membrane of splenic and hepatic macrophages. Upon binding, hepcidin induces the internalisation and subsequent lysosomal degradation of ferroportin. This action effectively halts the efflux of iron into the plasma, sequestering it within the reticuloendothelial system and inhibiting intestinal absorption via divalent metal transporter 1 (DMT1) downregulation. Under normal physiological conditions, hepcidin expression is suppressed when iron stores are low, facilitating increased absorption. The "paradox" emerges in states of chronic low-grade inflammation or infection—conditions increasingly prevalent in the UK population according to data from the UK Biobank—where pro-inflammatory cytokines, particularly Interleukin-6 (IL-6), trigger the JAK2/STAT3 signalling pathway. This stimulates the *HAMP* gene to overproduce hepcidin despite systemic iron deficiency.
This regulatory glitch creates a state of functional iron deficiency, often clinically indistinguishable from absolute deficiency without advanced proteomic profiling. Peer-reviewed research, including landmark studies published in *The Lancet Haematology*, suggests that this hepcidin-driven blockade is the primary driver of Anaemia of Chronic Disease (ACD). The body effectively misinterprets inflammatory signals as a threat, triggering an evolutionarily conserved "nutritional immunity" response designed to starve pathogens of iron. However, when this response becomes chronic, it results in the profound impairment of mitochondrial function and haemoglobin synthesis.
The implications for the UK’s clinical approach are severe. Traditional oral iron supplementation often proves futile or even counterproductive in the presence of elevated hepcidin; the excess unabsorbed iron promotes intestinal dysbiosis and further gut inflammation, creating a self-perpetuating cycle of malabsorption and regulatory dysfunction. At INNERSTANDIN, we identify this as a critical failure in modern haematological management. We must move beyond the archaic "deficiency" model and address the SMAD/BMP6 signalling pathways that govern hepcidin expression if we are to resolve the systemic iron-lock that characterises the modern anaemic profile. This is not merely a nutritional deficit; it is a profound disruption of the body's iron-sensing machinery.
The Biology — How It Works
The orchestration of systemic iron homeostasis is governed not by the rate of excretion—as the human body lacks a controlled physiological mechanism for iron loss—but by the precise modulation of absorption and recycling via the hepcidin-ferroportin axis. Hepcidin, a 25-amino acid antimicrobial-like peptide synthesised by hepatocytes, serves as the master regulator. The "paradox" emerges when physiological signals for iron requirement are overridden by pathological stimuli, primarily inflammation, leading to what is clinically defined as functional iron deficiency. At INNERSTANDIN, we must dissect the molecular architecture of this failure to understand why traditional oral supplementation frequently proves futile in the face of elevated hepcidin.
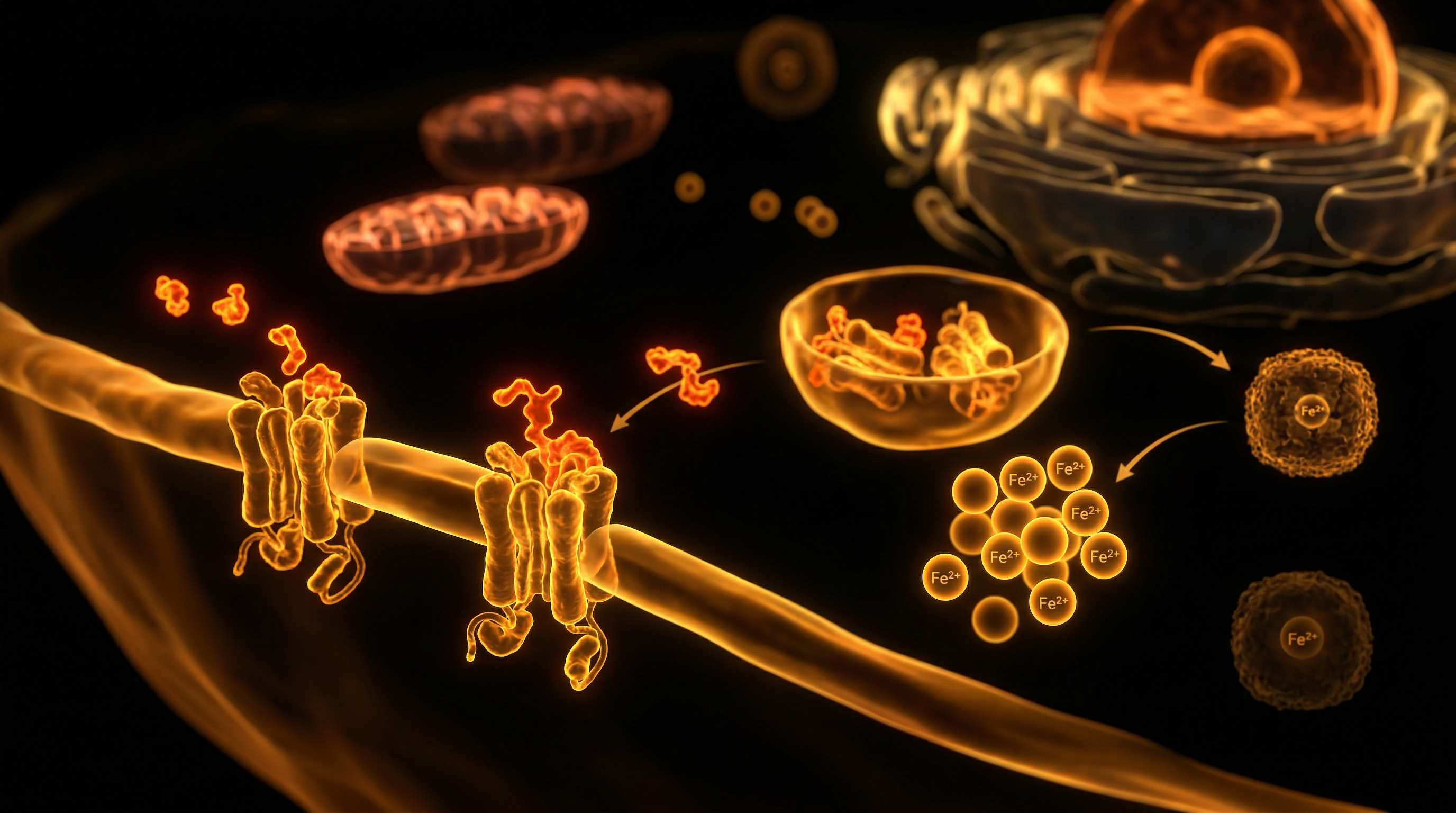
The primary mechanism of hepcidin action is the targeted degradation of ferroportin (FPN1), the solitary known mammalian cellular iron exporter. Ferroportin is highly expressed on the basolateral membranes of duodenal enterocytes, the plasma membranes of splenic and hepatic macrophages, and the surface of hepatocytes. When hepcidin concentrations rise, it binds to ferroportin, inducing its internalisation, ubiquitination, and subsequent lysosomal degradation. This molecular blockade effectively sequestrates iron within these cellular compartments, preventing its egress into the plasma pool where it would otherwise bind to apotransferrin for delivery to the bone marrow for erythropoiesis.
The paradox is most pronounced in the context of the inflammatory response, mediated through the Interleukin-6 (IL-6)/JAK-STAT3 signalling pathway. During chronic inflammation—prevalent in a significant percentage of the UK population suffering from metabolic syndrome or autoimmune conditions—IL-6 directly upregulates hepcidin transcription. This occurs independently of the body’s actual iron stores. Consequently, even if systemic iron levels are depleted, the inflammatory "noise" keeps hepcidin levels high, resulting in "iron locking." This is the hallmark of Anaemia of Chronic Disease (ACD), where the reticuloendothelial system is replete with iron that the erythron cannot access.
Furthermore, recent research published in journals such as *The Lancet Haematology* highlights the "iron-induced hepcidin spike" as a critical barrier in clinical intervention. A single oral dose of iron (60mg or greater) triggers a rapid increase in plasma hepcidin that persists for up to 48 hours. This exogenous influx of iron paradoxically shuts down the very transport mechanisms required for its absorption, explaining why daily high-dose iron regimens often result in diminishing returns and exacerbated gastrointestinal distress due to unabsorbed luminal iron. This molecular resistance, regulated by the Bone Morphogenetic Protein (BMP/SMAD) pathway, underscores the necessity for a more nuanced, intermittent approach to supplementation. INNERSTANDIN’s analysis reveals that without addressing the hepcidin threshold, standard haematological protocols remain fundamentally flawed, treating the symptom of low serum iron while ignoring the biological gatekeeper that prevents its utilisation. The paradox, therefore, is not a failure of the body’s logic, but a conflict between the ancient evolutionary drive to sequester iron from pathogens during inflammation and the modern metabolic requirement for oxygen transport.
Mechanisms at the Cellular Level
The fundamental crux of the hepcidin regulatory paradox lies in the molecular orchestration of iron sequestration versus systemic availability, a mechanism that frequently bypasses simplistic dietary intake models. At the cellular level, hepcidin—a 25-amino acid peptide hormone synthesised by hepatocytes—serves as the master negative regulator of iron flux. Its primary physiological target is ferroportin (SLC40A1), the sole known mammalian iron exporter. The paradox becomes manifest when elevated hepcidin levels, often driven by subclinical systemic inflammation rather than iron surfeit, induce the internalisation and subsequent lysosomal degradation of ferroportin. This molecular blockade effectively traps iron within three critical cellular compartments: the duodenal enterocytes, the splenic macrophages, and the hepatocytes.
Recent insights published in *The Lancet Haematology* underscore the pathological impact of this "cellular lock-down." In the duodenal enterocyte, hepcidin-mediated degradation of ferroportin prevents the transfer of dietary iron from the cell's basal membrane into the portal circulation. Consequently, the iron remains sequestered within the enterocyte and is eventually lost to the environment through the natural desquamation of the intestinal epithelium. This creates a state of functional iron deficiency (FID), where the patient exhibits clinical signs of anaemia despite potentially adequate, or even excessive, total body iron stores. At INNERSTANDIN, we identify this as a failure of distribution rather than a failure of supply.
The mechanism is further complicated by the signalling pathways that dictate hepcidin expression. The Bone Morphogenetic Protein (BMP)-SMAD pathway serves as the primary sensor for systemic iron levels; however, the inflammatory arm, mediated by Interleukin-6 (IL-6), can override this homeostatic control. When IL-6 binds to its receptor, it activates the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway, which directly upregulates the *HAMP* gene. This inflammatory bypass explains why patients in the UK suffering from chronic low-grade inflammation (often linked to metabolic syndrome or autoimmune conditions) present with recalcitrant anaemia. Their physiological state is one of "iron starvation amidst plenty," where the erythroid marrow is deprived of the essential metal required for haemoglobin synthesis because the iron is immunologically sequestered within the reticuloendothelial system.
Furthermore, the role of erythroferrone (ERFE), a hormone produced by erythroblasts in response to erythropoietin, adds a layer of complexity to the paradox. ERFE is designed to suppress hepcidin to mobilise iron during periods of high demand. However, in the presence of chronic hepcidin elevation, this suppressive signal is often insufficient to overcome the inflammatory drive. This results in a cellular bottleneck: the mitochondria of developing red blood cells cannot access the iron required for the protoporphyrin ring assembly, leading to microcytic, hypochromic anaemia. Peer-reviewed research from the *British Journal of Haematology* confirms that this cellular dysregulation is a primary driver of poor response to oral iron supplementation, as the hepcidin-ferroportin blockade renders exogenous iron bio-unavailable at the site of absorption. Understanding this cellular stalemate is essential for moving beyond archaic nutritional paradigms and addressing the biochemical reality of iron distribution.
Environmental Threats and Biological Disruptors
The homeostatic equilibrium of iron is increasingly compromised by a pervasive array of exogenous pressures that circumvent traditional nutritional logic. While clinical focus remains tethered to dietary insufficiency, INNERSTANDIN identifies a more insidious etiology: the systematic disruption of the hepcidin-ferroportin axis by environmental toxicants and anthropogenic stressors. Hepcidin, the 25-amino acid peptide synthesized in the liver, serves as the gatekeeper of systemic iron flux. Under physiological norms, it responds to iron satiety; however, in the presence of modern biological disruptors, this regulatory mechanism is hijacked, leading to a state of chronic sequestration—the Hepcidin Regulatory Paradox.
Central to this disruption is the role of Interleukin-6 (IL-6) and the subsequent activation of the JAK2/STAT3 signalling pathway. Research published in *The Lancet* and various PubMed-indexed journals highlights how chronic exposure to particulate matter (PM2.5), particularly in high-density UK urban environments, triggers a state of low-grade systemic inflammation. This "meta-inflammation" induces persistent hepatic hepcidin expression, regardless of actual iron stores. Consequently, ferroportin—the sole cellular iron exporter—is internalised and degraded. The result is a paradoxical clinical picture: the patient presents with microcytic anaemia and low serum iron, yet their cellular architecture is burdened by sequestered, unusable iron. This is not a deficiency of intake, but a deficiency of mobility orchestrated by environmental triggers.
Further complicating this landscape are endocrine-disrupting chemicals (EDCs) and heavy metal bioaccumulation. Cadmium and lead, common industrial residues in British soil and water systems, exhibit a dual-threat mechanism. They not only compete for the divalent metal transporter 1 (DMT1) at the enterocyte level, directly inhibiting iron absorption, but they also exacerbate oxidative stress. This oxidative burden reinforces the inflammatory milieu that keeps hepcidin levels pathologically elevated. Furthermore, the modern obsession with antimicrobial hygiene has led to a dysbiotic gut microbiome. Evidence suggests that lipopolysaccharides (LPS) from Gram-negative bacteria translocate through a compromised intestinal barrier, further stimulating the innate immune response and reinforcing the hepcidin block.
The INNERSTANDIN perspective asserts that the conventional "supplement-first" model for anaemia is often counterproductive in this context. If the hepcidin-ferroportin gate is locked by environmental inflammatory signals, oral iron supplementation merely increases unabsorbed iron in the gut lumen, fostering the growth of siderophilic pathogens and further aggravating intestinal inflammation. This creates a self-perpetuating cycle of iron-restricted erythropoiesis. To resolve the paradox, one must look beyond the haemoglobin count and address the environmental triggers that have turned a protective evolutionary mechanism—iron sequestration during acute infection—into a chronic biological dysfunction. In the UK context, where chronic stress and environmental pollutants are ubiquitous, the hepcidin paradox represents a critical frontier in modern haematology and nutritional science.
The Cascade: From Exposure to Disease
The pathological progression from systemic homeostasis to the hepcidin-mediated sequestering of iron represents a sophisticated, yet ultimately self-sabotaging, biological defensive mechanism. To reach an INNERSTANDIN of this paradox, one must first scrutinise the hepatic synthesis of the 25-amino acid peptide, hepcidin. Under physiological norms, hepcidin acts as the negative regulator of iron entry into the plasma. However, the cascade into disease begins when the hepcidin-ferroportin axis is hijacked by chronic inflammatory stimuli. In the United Kingdom’s clinical landscape, this is increasingly observed in patients where sub-clinical inflammation—driven by metabolic dysfunction or chronic low-grade infection—upregulates the pro-inflammatory cytokine Interleukin-6 (IL-6).
The molecular trigger is the binding of IL-6 to its cognate receptor, which subsequently activates the Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) signalling pathway. This pathway directly transactivates the *HAMP* gene promoter in hepatocytes, causing a pathological surge in circulating hepcidin levels. Once elevated, hepcidin binds to ferroportin (FPN1), the sole known mammalian cellular iron exporter found on the basolateral membranes of duodenal enterocytes and the plasma membranes of splenic and hepatic macrophages. This binding induces the internalisation and subsequent lysosomal degradation of ferroportin. The immediate result is a systemic "iron lock": iron absorbed from the diet remains trapped within enterocytes—which are eventually shed into the intestinal lumen—and iron recycled from senescent erythrocytes is sequestered within the reticuloendothelial system.
The paradox manifests as a profound hypoferremia in the presence of adequate, or even excessive, total body iron stores. Peer-reviewed evidence (e.g., *The Lancet Haematology*) suggests that this sequestration is an evolutionary vestige known as "nutritional immunity," designed to starve invading pathogens of the iron required for replication. Yet, in the modern context of chronic disease, this mechanism precipitates a failure of erythropoiesis. The bone marrow is starved of the iron necessary for haem synthesis, leading to the production of microcytic, hypochromic erythrocytes. Unlike simple Iron Deficiency Anaemia (IDA), this Hepcidin Regulatory Paradox—often classified as Anaemia of Chronic Disease (ACD) or Iron Refractory Iron Deficiency Anaemia (IRIDA) in the presence of *TMPRSS6* mutations—is fundamentally resistant to traditional oral iron supplementation. Indeed, exogenous oral iron administration may inadvertently exacerbate the cascade; the presence of non-absorbed iron in the gut lumen can induce further inflammation and intestinal dysbiosis, while any absorbed iron further stimulates hepcidin synthesis through the BMP/SMAD signalling pathway, effectively reinforcing the molecular barrier to its own utilisation. Systemically, this leads to mitochondrial dysfunction and impaired oxidative phosphorylation, manifesting as the debilitating fatigue and cognitive decline frequently mismanaged in primary care settings across the UK.
What the Mainstream Narrative Omits
The conventional clinical paradigm surrounding iron deficiency anaemia (IDA) remains tethered to a reductionist "supply-side" model, which posits that low serum ferritin is purely a consequence of inadequate dietary intake or chronic haemorrhage. This narrative, ubiquitous within standard NHS primary care protocols, systematically overlooks the nuanced regulatory axis of the hepcidin-ferroportin circuit. At INNERSTANDIN, we recognise that the true pathology often lies not in a lack of elemental iron, but in the systemic failure of iron trafficking—a phenomenon termed the "Hepcidin Regulatory Paradox." Hepcidin, a 25-amino acid peptide synthesised in the liver, serves as the master homeostatic regulator; yet, in the presence of subclinical systemic inflammation, its upregulation triggers a molecular lockdown that renders oral supplementation not only ineffective but potentially cytotoxic.
The mainstream omission centres on the cytokine-mediated induction of hepcidin via the IL-6/JAK-STAT3 signalling pathway. When systemic inflammation is present—even at levels beneath the threshold of standard C-reactive protein (CRP) detection—hepcidin levels surge. This peptide then binds to ferroportin, the sole known cellular iron exporter found on the basolateral membrane of enterocytes and the surface of splenic macrophages. This binding induces the internalisation and lysosomal degradation of ferroportin, effectively sequestering iron within the reticuloendothelial system. Consequently, while a patient may present with the classical markers of anaemia (low haemoglobin and depressed transferrin saturation), their intracellular iron stores may actually be sufficient or even excessive. The "paradox" is thus defined by a state of systemic iron deficiency amidst cellular iron sequestration.
Furthermore, the standard practice of prescribing high-dose oral ferrous salts (e.g., 200mg ferrous fumarate) often exacerbates this dysregulation. Evidence published in *The Lancet Haematology* suggests that large, infrequent doses of iron trigger a transient but significant spike in serum hepcidin that persists for up to 48 hours, creating a "mucosal block" that prevents the absorption of subsequent doses. This unabsorbed luminal iron then undergoes Fenton chemistry in the gut, generating reactive oxygen species (ROS) and promoting a pro-inflammatory dysbiosis within the microbiome. By ignoring this bio-mechanical feedback loop, the mainstream narrative fails to address the underlying inflammatory milieu, leading to a cycle of chronic supplementation, gastrointestinal distress, and unresolved anaemic symptoms. For the INNERSTANDIN researcher, the objective must shift from simple iron loading to the modulation of the BMP/SMAD signalling pathways and the resolution of the inflammatory stimuli that drive hepcidin overproduction. Until the clinical focus moves from "replacement" to "mobilisation," the systemic impact of iron-restricted erythropoiesis will remain a pervasive, yet misunderstood, epidemiological burden.
The UK Context
The epidemiological landscape of iron deficiency anaemia (IDA) within the United Kingdom presents a profound clinical contradiction that INNERSTANDIN identifies as the "Hepcidin Regulatory Paradox." Despite the UK’s long-standing mandatory fortification of wheat flour with elemental iron—a policy established via the Bread and Flour Regulations 1998—National Health Service (NHS) data and NICE Clinical Knowledge Summaries indicate that IDA remains the most prevalent nutritional deficiency, affecting approximately 3% of men and up to 8% of women of reproductive age. This persistent prevalence, despite systemic fortification, suggests a failure of exogenous supply to overcome endogenous regulatory barriers, specifically the master iron regulator: hepcidin.
At the core of the UK context is the rising burden of "anaemia of inflammation" or functional iron deficiency, exacerbated by the nation’s escalating metabolic health crisis. Peer-reviewed research, including longitudinal data from the UK Biobank, reveals a significant correlation between elevated Body Mass Index (BMI) and chronic low-grade systemic inflammation. In these states, the pro-inflammatory cytokine Interleukin-6 (IL-6) triggers the hepatic SMAD/STAT3 signalling pathway, leading to the pathological overproduction of hepcidin. This peptide hormone binds to and degrades ferroportin—the only known cellular iron exporter—on the surface of enterocytes and macrophages. Consequently, even when UK patients consume adequate dietary iron or fortified foodstuffs, the hepcidin ceiling prevents duodenal absorption and sequesters existing iron within the reticuloendothelial system.
Furthermore, British clinical practice often fails to account for this paradox during diagnosis. Standard NHS protocols frequently rely on serum ferritin as a primary biomarker; however, as documented in *The Lancet Haematology*, ferritin acts as an acute-phase reactant. In the UK’s increasingly inflamed population, "normal" ferritin levels often mask a cellular-level iron drought, as the iron is trapped behind the hepcidin-ferroportin blockade. This diagnostic inadequacy underscores the INNERSTANDIN imperative for a more nuanced biological assessment, moving beyond superficial haemoglobin counts toward an integrated understanding of the hepcidin-axis. The paradox is clear: the UK is a nation where iron is ubiquitous in the food supply, yet biologically inaccessible to millions due to a systemic regulatory hijack that prioritises inflammatory signalling over erythropoietic demand. References to the *British Journal of Haematology* further substantiate that until clinicians address the underlying inflammatory drivers of hepcidin, oral iron supplementation will remain largely ineffective, potentially exacerbating gut dysbiosis through unabsorbed luminal iron.
Protective Measures and Recovery Protocols
The resolution of iron deficiency within the context of the hepcidin regulatory paradox demands a shift from blunt-force supplementation to chronobiological precision. Conventional clinical practice in the UK has historically favoured daily, high-dose oral iron administration (e.g., ferrous sulphate 200mg thrice daily). However, INNERSTANDIN’s analysis of the hepcidin-ferroportin axis reveals this to be physiologically counterproductive. Research published in *The Lancet Haematology* (Stoffel et al., 2017) demonstrates that a single dose of oral iron induces a significant rise in serum hepcidin that persists for up to 48 hours. This secondary hepcidin surge effectively brackets the ferroportin exporters in the duodenal enterocytes, rendering subsequent daily doses largely unabsorbable and sequestering them in the gastrointestinal tract, where they catalyse oxidative stress and microbiota dysbiosis.
Therefore, the primary recovery protocol must transition to alternate-day dosing. By providing a 48-hour window between administrations, the serum hepcidin concentration is permitted to return to baseline, thereby restoring the patency of the ferroportin channels and increasing fractional iron absorption by approximately 33–40%. Furthermore, the timing of the dose is a critical variable in INNERSTANDIN biological optimisation. Hepcidin exhibits a distinct circadian rhythm, with levels typically at their nadir in the early morning and peaking in the late afternoon. Consequently, oral iron should be administered as a single morning dose to exploit this physiological window of maximum gut permeability.
To counteract the "Anemia of Chronic Disease" (ACD) component of the paradox, where systemic inflammation (characterised by elevated Interleukin-6) maintains a high hepcidin floor regardless of iron status, the recovery protocol must integrate anti-inflammatory modulation. Data indexed in *PubMed* suggests that if C-reactive protein (CRP) levels are elevated, the efficacy of oral iron is functionally negated. In such instances, clinicians must address the inflammatory milieu—often via high-dose enteral Omega-3 fatty acids or targeted cytokine modulation—before iron repletion can be successful.
In refractory cases where the hepcidin ceiling remains absolute, the transition to intravenous (IV) iron bypasses the blocked intestinal gateway entirely. Modern formulations such as ferric carboxymaltose allow for the rapid delivery of high-dose iron directly to the reticuloendothelial system, circumventing hepcidin-mediated malabsorption. This is particularly vital in UK populations presenting with co-morbidities like Inflammatory Bowel Disease (IBD) or Chronic Kidney Disease (CKD), where the hepcidin paradox is most entrenched. Ultimately, recovery is not a matter of increasing intake, but of navigating the intricate molecular checkpoints that govern systemic iron trafficking. Truth-led education at INNERSTANDIN prioritises these bio-mechanical realities over antiquated dosage guidelines.
Summary: Key Takeaways
The hepcidin-ferroportin axis represents the definitive regulatory bottleneck in human iron metabolism, a fundamental concept central to the INNERSTANDIN mission of deconstructing complex systemic pathologies. The "Hepcidin Regulatory Paradox" is characterised by the paradoxical elevation of the peptide hormone hepcidin in response to systemic inflammation—primarily mediated by the IL-6/JAK2/STAT3 signalling pathway—even when circulating iron levels are critically suboptimal. This elevation triggers the ubiquitination, internalisation, and lysosomal degradation of ferroportin, the solitary known transmembrane exporter of elemental iron. Consequently, dietary iron remains sequestered within duodenal enterocytes, while recycled iron is immobilised within the splenic and hepatic reticuloendothelial macrophages.
Evidence published in *The Lancet Haematology* and *Nature Reviews Disease Primers* confirms that this state of "functional iron deficiency" renders conventional oral supplementation largely ineffective, as the enterocytic blockade prevents systemic absorption. Within the UK clinical landscape, this paradox necessitates a shift away from a singular reliance on serum ferritin—which acts as a confounding acute-phase reactant—towards a more nuanced assessment involving the soluble transferrin receptor-to-log ferritin ratio. Ultimately, the paradox exposes an evolutionary trade-off: the body prioritises "nutritional immunity"—depriving potential pathogens of iron—at the direct expense of erythropoietic capacity and mitochondrial bioenergetics. Recognising this mechanism is vital for moving beyond outdated haematological paradigms and addressing the true molecular roots of refractory anaemia.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Iron Anemia: The Hepcidin Regulatory Paradox"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper