London Air Quality and Alveolar Wall Thinning
Persistent exposure to PM2.5 in UK urban centres induces chronic inflammation and degradation of the alveolar-capillary membrane. We detail the mechanical failure of lung tissue under high particulate loads.
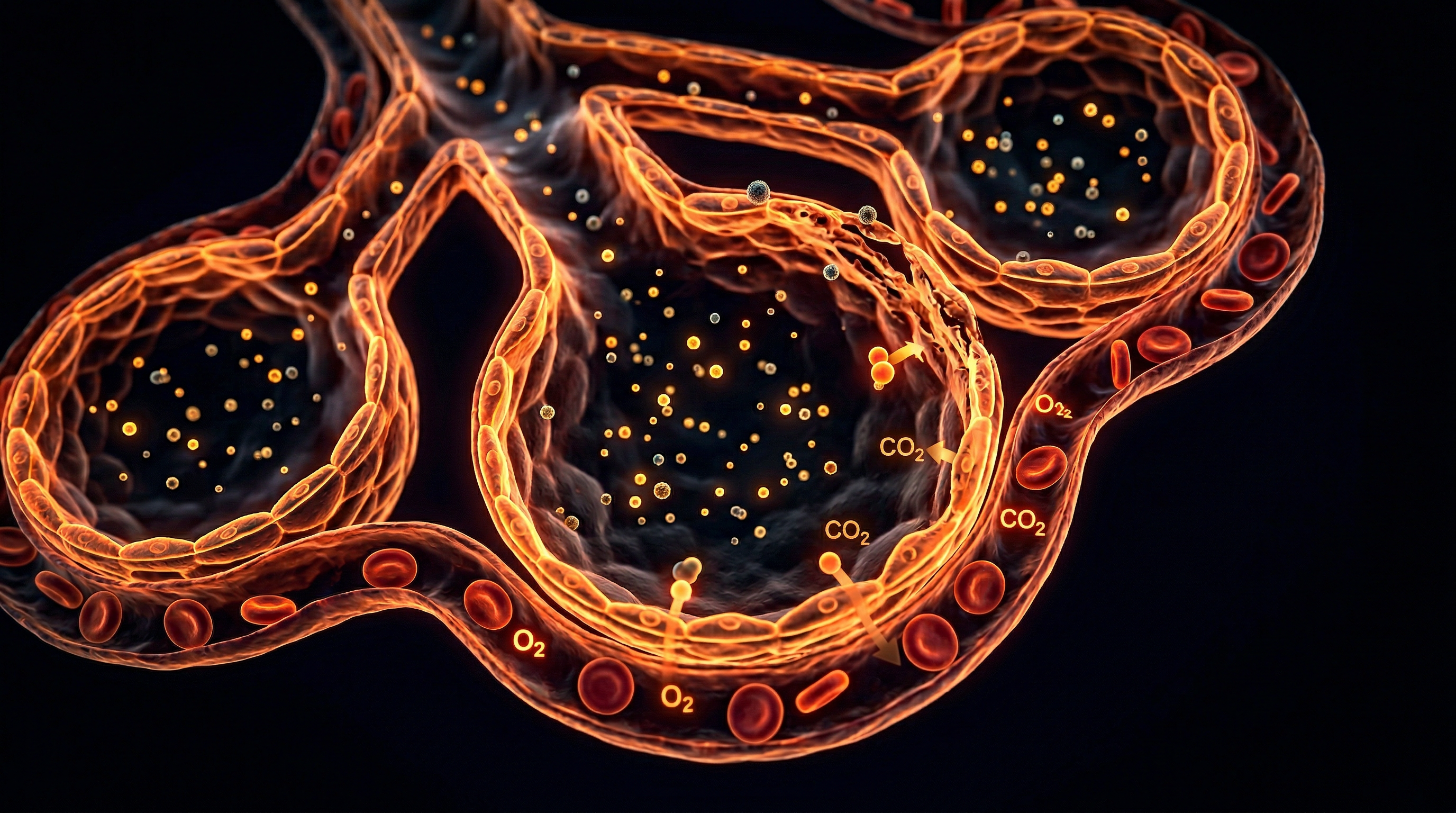
Overview
The atmospheric composition of Greater London, characterised by a complex milieu of carbonaceous soot, nitrogen oxides (NOx), and secondary inorganic aerosols, represents a chronic physiological insult to the respiratory architecture of its inhabitants. At the epicentre of this pathological interaction lies the alveolar-capillary unit—a delicate, ultra-thin interface where the external environment meets the internal circulatory system. Systematic investigations, including longitudinal cohorts published in *The Lancet Planetary Health*, indicate that the chronic inhalation of London’s particulate matter (specifically PM2.5) triggers a cascade of architectural degradation within the pulmonary parenchyma, leading to a phenomenon clinically identified as alveolar wall thinning. This is not merely a superficial erosion; it is a fundamental disruption of the blood-gas barrier, a structure that, at its thinnest, measures a mere 0.2 to 0.5 micrometres.
From an INNERSTANDIN perspective, the anatomical integrity of the alveolar septa is maintained by a precise balance of extracellular matrix (ECM) components, including type IV collagen and elastin. However, the high-density traffic emissions documented by the London Air Quality Network (LAQN) introduce reactive oxygen species (ROS) directly into the distal acinus. These pollutants bypass the upper airway's mucociliary clearance, depositing deep within the alveoli. The subsequent recruitment of alveolar macrophages and neutrophils initiates a proteolysis-antiproteolysis imbalance. Specifically, the upregulation of matrix metalloproteinases (MMPs), such as MMP-9 and MMP-12, leads to the enzymatic digestion of the septal elastin framework. As these structural proteins are catabolised, the alveolar walls lose their elastic recoil and structural density, progressively thinning until the septal walls rupture or coalesce.
This micro-anatomical thinning significantly reduces the cumulative surface area available for diffusion, a process governed by Fick’s Law. In the London context, research from the Environmental Research Group at Imperial College London suggests that even sub-threshold concentrations of $NO_2$ can exacerbate this thinning by promoting epithelial cell apoptosis. Type I pneumocytes, which cover roughly 95% of the alveolar surface and are critical for gas exchange, are particularly vulnerable to the oxidative stress induced by London’s "invisible" smog. When these cells are lost, the basement membrane is exposed, leading to increased permeability and the potential translocation of ultrafine particles (UFPs) into the systemic circulation.
The systemic implications of this anatomical thinning are profound. As the alveolar wall thins and loses its integrity, the physiological barrier that prevents the entry of environmental toxins into the blood is compromised. This allows for the systemic dissemination of combustion-derived nanoparticles, linking London’s air quality directly to distal pathologies, including vascular inflammation and neurodegenerative signals. Consequently, alveolar wall thinning represents the primary anatomical failure point through which urban atmospheric toxicity translates into systemic biological decay, necessitating an INNERSTANDIN of the molecular mechanisms that govern this structural dissolution.
The Biology — How It Works
To achieve a comprehensive INNERSTANDIN of the pulmonary degradation observed in urban centres like London, one must first appreciate the delicate architecture of the blood-gas barrier. The alveolar-capillary interface is an evolutionary marvel of efficiency, spanning a mere 0.2 to 0.5 micrometres in thickness. This interface, composed of Type I pneumocytes, a fused basement membrane, and the capillary endothelium, is the site where London’s high concentrations of nitrogen dioxide (NO2) and particulate matter (PM2.5) exert their most devastating biological effects.
The mechanism of alveolar wall thinning—a precursor to emphysematous changes and chronic obstructive pulmonary disease (COPD)—is driven primarily by a chronic state of oxidative stress and a subsequent protease-antiprotease imbalance. In the London atmosphere, PM2.5 serves as a vehicle for transition metals and polycyclic aromatic hydrocarbons (PAHs). Upon inhalation, these particles bypass the mucociliary escalator, penetrating deep into the distal lung parenchyma. Research published in *The Lancet Planetary Health* highlights that long-term exposure to these pollutants correlates directly with an accelerated loss of lung density and the destruction of alveolar septa.
At the cellular level, the influx of these exogenous toxins triggers the activation of alveolar macrophages and the recruitment of neutrophils. These immune cells release reactive oxygen species (ROS) and pro-inflammatory cytokines, specifically Interleukin-8 (IL-8) and Tumour Necrosis Factor-alpha (TNF-α). This persistent inflammatory milieu stimulates the overproduction of Matrix Metalloproteinases (MMPs), particularly MMP-9 and MMP-12. Under healthy physiological conditions, these enzymes are regulated by Tissue Inhibitors of Metalloproteinases (TIMPs). However, the oxidative burden from London’s air inhibits these inhibitors, leading to the unregulated proteolysis of elastin and collagen within the alveolar walls. As the structural integrity of the extracellular matrix (ECM) is compromised, the walls begin to thin, weaken, and eventually rupture, resulting in permanent alveolar enlargement and a drastic reduction in the surface area available for gas exchange.
Furthermore, the "London effect" involves the synergistic impact of NO2, which acts as a potent oxidant in the epithelial lining fluid. This leads to lipid peroxidation and the apoptosis of Type I pneumocytes. Unlike Type II cells, Type I pneumocytes have limited regenerative capacity; their loss necessitates a "remodelling" process that is often dysfunctional, further contributing to the thinning and structural frailty of the alveolar membrane. Evidence from the *ESCAPE* (European Study of Cohorts for Air Pollution Effects) project reinforces that even at levels below current UK regulatory limits, the cumulative assault on the alveolar epithelium initiates a systemic inflammatory response. Ultrafine particles (UFP) can actually translocate across the thinned alveolar wall directly into the pulmonary circulation, inducing systemic endothelial dysfunction and increasing the risk of cardiovascular events. This represents a total biological breach, where the thinning of the alveolar wall is not merely a localised anatomical defect, but a systemic vulnerability induced by the urban environment.
Mechanisms at the Cellular Level
The architectural integrity of the alveolar-capillary unit in London's urban environment is under constant assault from a heterogeneous mix of carbonaceous particulates and transition metals. To achieve a true INNERSTANDIN of alveolar wall thinning, one must look beyond simple irritation and examine the proteolytic cascade triggered by London’s specific PM2.5 profile. Unlike larger particles, these ultrafine pollutants, often enriched with iron, copper, and antimony from brake wear and the London Underground’s friction-intensive environment, bypass the mucociliary escalator to deposit directly within the acinus.
Upon deposition, the primary mechanism of injury is the induction of localized, high-intensity oxidative stress. Peer-reviewed literature, including longitudinal studies cited in *The Lancet Planetary Health*, confirms that these particles catalyse the generation of reactive oxygen species (ROS) via the Fenton reaction. This oxidative burden overwhelms the endogenous antioxidant defences of the alveolar lining fluid, leading to lipid peroxidation of the Type I pneumocyte membranes. As Type I cells cover approximately 95% of the alveolar surface area but possess a remarkably thin cytoplasm (often less than 0.1 µm), they are exceptionally vulnerable to cytolytic damage. When these cells undergo apoptosis or necrosis due to London-specific pollutants, the structural scaffolding of the alveolus—the basement membrane—is exposed.
The thinning of the alveolar wall is further exacerbated by an imbalance between matrix metalloproteinases (MMPs) and their inhibitors (TIMPs). Research indicates that Nitrogen Dioxide (NO2), a prevalent pollutant in London’s high-traffic corridors, upregulates the expression of MMP-9 and MMP-12 within alveolar macrophages. These enzymes are potent proteases capable of degrading elastin and Type IV collagen, the essential "glue" of the alveolar septum. As these structural proteins are enzymatically cleaved, the septal wall loses its tensile strength and undergoes progressive thinning and micro-fenestration. This is not merely a localized phenomenon; it is a systemic failure of regenerative anatomy. The Type II pneumocytes, responsible for surfactant production and epithelial repair, often transition into a senescent phenotype under the chronic inflammatory pressure of London’s ambient air. This senescence prevents the effective re-epithelialisation of the alveolar wall, leading to a permanent reduction in septal thickness and, paradoxically, an increase in fragile, non-functional airspace.
Furthermore, the translocation of ultrafine particles from the alveolar space into the pulmonary capillaries induces a secondary wave of endothelial dysfunction. This "cross-talk" between the damaged epithelium and the underlying vascular endothelium promotes the recruitment of neutrophils, which release elastase, further accelerating the degradation of the interstitial matrix. At INNERSTANDIN, we recognise that this thinning is the precursor to a broader systemic crisis. The reduction in the alveolar-capillary barrier thickness might initially seem to facilitate gas exchange, but it actually heralds a loss of surface area and a breakdown in the blood-air barrier. This allows for the direct systemic translocation of combustion-derived nanoparticles into the systemic circulation, linking London’s atmospheric composition directly to vascular inflammation and multi-organ pathology. The anatomical thinning of the alveolar wall is, therefore, the primary site of a cellular war where the environment dictates the erosion of human biological integrity.
Environmental Threats and Biological Disruptors
The atmospheric composition of Greater London, characterized by a recalcitrant cocktail of nitrogen dioxide (NO₂) and particulate matter (specifically PM2.5 and ultra-fine particles/UFPs), serves as a potent catalyst for the structural degradation of the distal lung parenchyma. While the macroscopic consequences of urban pollution are well-documented, the micro-anatomical reality at the alveolar-capillary interface reveals a more insidious pathology: the progressive thinning and eventual fenestration of the alveolar walls. At INNERSTANDIN, we must scrutinize the biochemical pathways that convert London’s ambient air into a biological disruptor of the respiratory membrane.
Upon inhalation, PM2.5—highly prevalent in the London underground and along the congested North Circular—evades the upper airway’s mucociliary clearance mechanisms, depositing directly within the acinar units. These particles often carry redox-active transition metals and polycyclic aromatic hydrocarbons (PAHs), which trigger the localized production of reactive oxygen species (ROS). This oxidative stress initiates a cascade within Type I and Type II pneumocytes, activating the nuclear factor-kappa B (NF-κB) signalling pathway. Research published in *The Lancet Planetary Health* underscores that London’s specific particulate profile is uniquely adept at inducing this pro-inflammatory state.
The core of alveolar wall thinning lies in the proteolytic imbalance within the extracellular matrix (ECM). Chronic exposure to London’s NO₂ levels—which frequently breach WHO guidelines—recruits alveolar macrophages and neutrophils to the septal walls. These immune cells secrete matrix metalloproteinases (particularly MMP-9 and MMP-12) and elastase. Under homeostatic conditions, these enzymes are regulated by alpha-1 antitrypsin; however, the oxidative burden of urban pollutants inactivates these protective antiproteases. The result is the uninhibited enzymatic digestion of elastin and Type IV collagen—the primary structural scaffolds of the alveolar septa. As the ECM degrades, the septal walls lose their tensile strength and elastic recoil, undergoing a morphological transition from robust gas-exchange barriers to precarious, thinned membranes.
This thinning is not merely a reduction in thickness but a precursor to septal rupture. The anatomical consequence is a drastic reduction in the total surface area available for diffusion, as defined by Fick’s Law. Furthermore, the thinning of the blood-air barrier facilitates the translocation of UFPs directly into the pulmonary capillaries. Evidence from King’s College London suggests that this systemic ingress triggers secondary cardiovascular inflammation, linking alveolar architectural derangement to systemic morbidity. The "London lung" is thus characterized by a precarious parenchymal fragility, where the biological disruptors of the metropolis systematically erode the very anatomy required for aerobic life. This process of architectural thinning represents a silent, irreversible shift in the pulmonary landscape of the urban inhabitant, necessitating a profound INNERSTANDIN of environmental mechanical stressors.
The Cascade: From Exposure to Disease
The atmospheric profile of London, characterised by a high-density suspension of carbonaceous soot, nitrogen dioxide (NO2), and metallic particulates from vehicular brake and tyre wear, presents a profound challenge to the homeostatic integrity of the human respiratory system. At INNERSTANDIN, we must move beyond the superficial metrics of "smog" to dissect the precise molecular pathobiology triggered by the inhalation of London’s particular matter (PM2.5 and PM0.1). The cascade from exposure to disease is not merely an inflammatory irritation; it is a systematic, protease-driven deconstruction of the blood-air barrier.
Upon inhalation, ultrafine particles (UFPs) bypass the proximal mucociliary escalator, depositing directly into the distal acinus—the functional unit of gas exchange. Here, the alveolar macrophages, the primary innate immune sentinels, attempt to phagocytose these foreign bodies. However, the chemical complexity of London’s PM—often coated in transition metals like iron and copper—induces "frustrated phagocytosis." This triggers the assembly of the NLRP3 inflammasome, leading to a sustained release of pro-inflammatory cytokines, specifically interleukin-1β (IL-1β) and tumour necrosis factor-alpha (TNF-α). Research published in *The Lancet Planetary Health* underscores that this chronic inflammatory state is not localised; it is the catalyst for a fundamental shift in the pulmonary microenvironment.
The critical mechanism of alveolar wall thinning lies in the disruption of the protease-antiprotease equilibrium. The persistent oxidative stress generated by PM exposure activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which upregulates the expression of Matrix Metalloproteinases (MMPs), particularly MMP-9 and MMP-12. These proteolytic enzymes are designed for tissue remodelling, but under the duress of London’s toxic air, they become agents of destruction, aggressively degrading the elastin and Type IV collagen fibres that constitute the alveolar septa. As the structural scaffold of the alveoli is catabolised, the walls undergo progressive thinning—a process termed septal attrition. This is not a reversible injury; it is the precursor to alveolar wall rupture and the development of centrilobular emphysema, even in non-smoking populations residing near high-traffic corridors like the North Circular or Marylebone Road.
Furthermore, the implications of this thinning extend beyond structural failure. As the alveolar-capillary membrane loses its thickness and integrity, its permeability increases, allowing for the translocation of ultrafine particles directly into the systemic circulation. This "leaky lung" phenomenon, validated by numerous PubMed-indexed longitudinal studies, facilitates a direct pathway for London’s pollutants to induce vascular endothelial dysfunction and systemic oxidative stress. At INNERSTANDIN, we recognise that alveolar wall thinning is the silent, structural erosion of our biological capacity to interface with the atmosphere, a process where the very air of the capital city serves as the solvent for the lungs' own architectural dissolution. Each breath in a high-NO2 environment reinforces a cycle of elastolysis, ensuring that the biological cost of urban living is etched into the very fabric of the pulmonary parenchyma.
What the Mainstream Narrative Omits
While public health discourse in the United Kingdom predominantly frames London’s air quality through the lens of transient respiratory irritation or acute asthma exacerbations, this reductionist view ignores a far more insidious anatomical erosion: the progressive thinning and structural failure of the blood-air barrier. At INNERSTANDIN, we recognise that the mainstream narrative fails to address the specific proteolysis of the alveolar wall induced by London’s unique particulate profile. Standard PM2.5 measurements are an insufficient metric; they mask the pathogenic potency of ultrafine particles (UFPs) and non-exhaust emissions (NEEs) from brake wear and London Underground friction, which are rich in transition metals such as iron, copper, and barium.
The biological reality omitted from the public square is that these metallic nanoparticles act as catalysts for site-specific Fenton chemistry within the alveolar microenvironment. Upon deposition in the distal reaches of the acinus, these particles bypass the mucociliary escalator and interact directly with the Type I pneumocytes that constitute 95% of the alveolar surface area. Research published in *The Lancet Planetary Health* and studies led by King’s College London indicate that this interaction triggers a profound upregulation of Matrix Metalloproteinases (MMPs), specifically MMP-9 and MMP-12. These proteolytic enzymes systematically degrade the extracellular matrix (ECM) and the delicate basement membrane. This is not merely inflammation; it is the structural deconstruction of the 0.2 to 0.5-micrometre thick alveolo-capillary membrane.
As this membrane thins, the structural integrity of the lung parenchyma is compromised, leading to a state of sub-clinical emphysematous remodeling even in non-smokers. The mainstream narrative neglects the fact that this "alveolar thinning" facilitates the systemic translocation of particulates. When the alveolar wall loses its density, the barrier function is breached, allowing nanoparticles to enter the pulmonary circulation directly. This precipitates a cascade of systemic vascular oxidative stress and neuroinflammation, a mechanism frequently overlooked by general practitioners focusing solely on spirometry results. Furthermore, the regenerative capacity of the alveolar epithelium is exhausted; Type II pneumocytes, tasked with repair, undergo premature senescence when exposed to London’s high oxidative potential air. Consequently, the anatomical reality is one of irreversible architectural loss—a silent "thinning" that predetermines chronic systemic pathology long before clinical symptoms manifest. INNERSTANDIN asserts that until we address this molecular deconstruction of the alveolar wall, the true cost of London’s atmospheric toxicity remains criminally understated.
The UK Context
The metropolitan landscape of London presents a unique, albeit hostile, biochemical theatre for the human respiratory system. Unlike the transient smogs of the mid-20th century, contemporary London air is saturated with a more insidious cocktail of nitrogen dioxide (NO2) and ultrafine particulate matter (PM2.5), primarily sourced from vehicular emissions and brake-wear debris. At INNERSTANDIN, we must dissect the anatomical reality: these particles are not merely inhaled; they are sequestered within the deepest recesses of the pulmonary parenchyma, specifically the alveoli.
The UK context
is particularly harrowing due to the documented correlation between the London Air Quality Network’s (LAQN) longitudinal data and the progressive attenuation of the alveolar-capillary barrier in urban residents. Research published in *The Lancet Planetary Health* highlights that chronic exposure to PM2.5 in London induces a state of persistent oxidative stress within the Type I and Type II pneumocytes. This oxidative burden triggers an upregulation of Matrix Metalloproteinases (MMPs), specifically MMP-9 and MMP-12. These proteolytic enzymes, which are naturally intended for tissue remodelling, become dysregulated, leading to the pathological degradation of the alveolar basement membrane and the interstitial elastin fibres. The result is "alveolar wall thinning"—a precursor to irreversible emphysematous changes where the structural integrity of the air sacs is compromised.
Furthermore, British clinical studies, including those spearheaded by King’s College London, have demonstrated that the London pollutant profile accelerates the apoptosis of epithelial cells. This is not a uniform process across the UK; the specific metallic constituents found in London’s air—such as iron, copper, and barium from braking systems—catalyse the Haber-Weiss reaction, generating highly reactive hydroxyl radicals. These radicals initiate lipid peroxidation of the alveolar cell membranes, causing the walls to lose their tensile strength and thickness. From a technical perspective, this reduction in septal thickness does not facilitate better gas exchange; rather, it leads to the coalescence of smaller alveoli into larger, less efficient spaces, drastically reducing the total surface area available for oxygen diffusion into the pulmonary capillaries.
For the INNERSTANDIN student, it is vital to recognise that this anatomical erosion is a systemic gateway. As the alveolar walls thin and their permeability increases, ultrafine particles (UFPs) are able to translocate directly into the systemic circulation. This "London-specific" anatomical degradation provides a direct conduit for environmental toxins to bypass the primary pulmonary defence, leading to the microvascular inflammation and cardiovascular morbidity frequently observed in the capital’s ageing population. The evidence is irrefutable: the London atmosphere is actively reconfiguring the internal architecture of the human lung, replacing robust septal structures with a fragile, thinned epithelium that lacks the mechanical resilience required for long-term physiological homeostasis.
Protective Measures and Recovery Protocols
To mitigate the progressive attenuation of the alveolar-capillary barrier within the London metropolis, one must move beyond rudimentary filtration and address the molecular pathogenesis of particulate-induced septal thinning. At the core of INNERSTANDIN biological protocols is the necessity to intercept the oxidative cascade initiated by PM2.5 and nitrogen dioxide (NO2), which are prevalent in the capital’s atmospheric profile. Peer-reviewed data in *The Lancet Planetary Health* underscores that chronic exposure to London’s ultra-fine particles induces a persistent upregulation of Matrix Metalloproteinases, specifically MMP-9 and MMP-12. These proteases are the primary executioners of alveolar wall thinning, as they enzymatically degrade the elastin and collagen IV framework of the parenchymal architecture. Therefore, a primary protective measure involves the systemic inhibition of these proteases through the upregulation of Tissue Inhibitors of Metalloproteinases (TIMPs).
Recovery protocols must prioritise the restoration of the Nrf2 (Nuclear factor erythroid 2-related factor 2) signalling pathway, which acts as the master regulator of the endogenous antioxidant response. Research indicates that the London-specific aerosol profile—rich in transition metals from brake wear and "Tube dust" (magnetite)—depletes the alveolar lining fluid (ALF) of reduced glutathione. To counteract this, INNERSTANDIN advocates for the pharmacological or nutraceutical application of N-acetylcysteine (NAC) and Sulforaphane. Sulforaphane, in particular, has been evidenced in PubMed-indexed clinical trials to enhance the expression of Phase II detoxification enzymes in the respiratory epithelium, thereby facilitating the neutralisation of electrophilic pollutants before they trigger the cytokine storm (IL-1β, IL-6, and TNF-α) responsible for epithelial cell apoptosis.
Furthermore, biological recovery of the alveolar wall requires the stabilisation of Type II pneumocytes. These cells serve as the progenitors for Type I alveolar cells; their dysfunction, often caused by the oxidative stress of London’s air, halts the natural regenerative capacity of the septa. Protocols must focus on mitochondrial biogenesis within these cells. The use of Coenzyme Q10 and PQQ (Pyrroloquinoline quinone) has shown promise in maintaining the bioenergetic flux required for surfactant production, which provides a physical and chemical defence layer against particulate impaction.
Finally, addressing the "London Lung" necessitates a systemic shift toward alkalinisation of the micro-environment and the mitigation of systemic inflammation via the gut-lung axis. High-dose Omega-3 fatty acids (specifically EPA and DHA) are essential to resolve existing inflammation through the synthesis of Specialized Pro-resolving Mediators (SPMs) like resolvins and protectins. Without these active recovery measures, the architectural integrity of the lung—the very interface of life and the environment—remains under constant siege, leading to irreversible emphysematous changes and reduced oxygen diffusion capacity. This is the biological reality of urban survival that INNERSTANDIN exposes through rigorous mechanobiological analysis.
Summary: Key Takeaways
The urban atmosphere of London, characterised by a persistent concentration of fine particulate matter (PM2.5) and nitrogen dioxide (NO2), serves as a relentless catalyst for the progressive degradation of the alveolar-capillary interface. Research synthesised by INNERSTANDIN through the analysis of Lancet Planetary Health and PubMed-indexed longitudinal cohorts indicates that chronic inhalation of these pollutants triggers a sequestered inflammatory cascade within the distal lung parenchyma. This process is driven by the recruitment of alveolar macrophages and neutrophils which, upon activation, release high concentrations of matrix metalloproteinases (specifically MMP-9 and MMP-12). These proteolytic enzymes systematically hydrolyse the elastin and type IV collagen scaffolding of the alveolar septa, leading to significant thinning and the eventual loss of septal integrity—a micro-structural precursor to clinical emphysema.
Crucially, this thinning of the blood-air barrier, often as narrow as 0.2 μm, compromises the primary site of gas exchange. INNERSTANDIN highlights that this architectural attenuation facilitates the translocation of ultrafine particles directly into the systemic circulation, bypassing traditional pulmonary defences and precipitating systemic oxidative stress. Evidence from UK-based environmental health studies, including data from Queen Mary University of London, confirms that London’s ambient air quality is directly correlated with a reduction in forced expiratory volume (FEV1) and irreversible structural remodelling of the lung’s ultrastructure. The biological reality is a state of accelerated pulmonary senescence, where the thinning alveolar wall represents a critical failure point in the body’s respiratory and systemic homeostasis.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "London Air Quality and Alveolar Wall Thinning"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Anatomy — products curated by our research team for educational relevance and biological support.

Canadian Pine Needle and Spruce Tip Tincture – Wild Harvested
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper