Lp(a): Britain’s Silent Genetic Killer
Lipoprotein (a) is a highly inflammatory genetic factor that remains largely ignored by standard UK lipid panels. Learn how this specific particle drives premature atherosclerosis regardless of a healthy lifestyle.
# Lp(a): Britain’s Silent Genetic Killer
Overview
In the clinical landscape of British cardiovascular medicine, a phantom haunts the wards of the NHS. It is a molecule that strikes with surgical precision, often claiming the lives of those who appear to be the "picture of health"—the marathon runners, the lifelong vegetarians, and the middle-aged professionals with seemingly perfect blood pressure. This phantom is Lipoprotein (a), commonly referred to as Lp(a) (pronounced "LP-little-a").
For decades, the mainstream medical narrative has focused almost exclusively on Low-Density Lipoprotein (LDL) cholesterol—the so-called "bad" cholesterol. Patients are told that if they lower their LDL through statins and a low-fat diet, they are safe from the ravages of heart disease. This is a half-truth that masks a more sinister reality. While LDL is indeed a factor in atherosclerosis, Lp(a) is a far more potent, inflammatory, and thrombogenic particle that remains entirely untouched by standard lifestyle interventions and traditional statin therapy.
In the United Kingdom, it is estimated that approximately one in five people (roughly 12 million individuals) possess elevated levels of Lp(a) due to their genetic makeup. Despite this, Lp(a) is not part of the standard lipid panel performed at GP surgeries. It is a "silent killer" because most people—and many frontline clinicians—are unaware of its existence until a catastrophic event, such as a premature myocardial infarction or stroke, occurs.
This article serves as a comprehensive exposé on the biology of Lp(a), the mechanisms by which it destroys the vascular system, and why the current UK healthcare framework has failed to address this genetic ticking time bomb.
Key Fact: High levels of Lp(a) are 100% genetically determined. Unlike LDL, Lp(a) levels do not fluctuate significantly based on diet, exercise, or smoking habits. You are born with your Lp(a) trajectory.
---
The Biology — How It Works
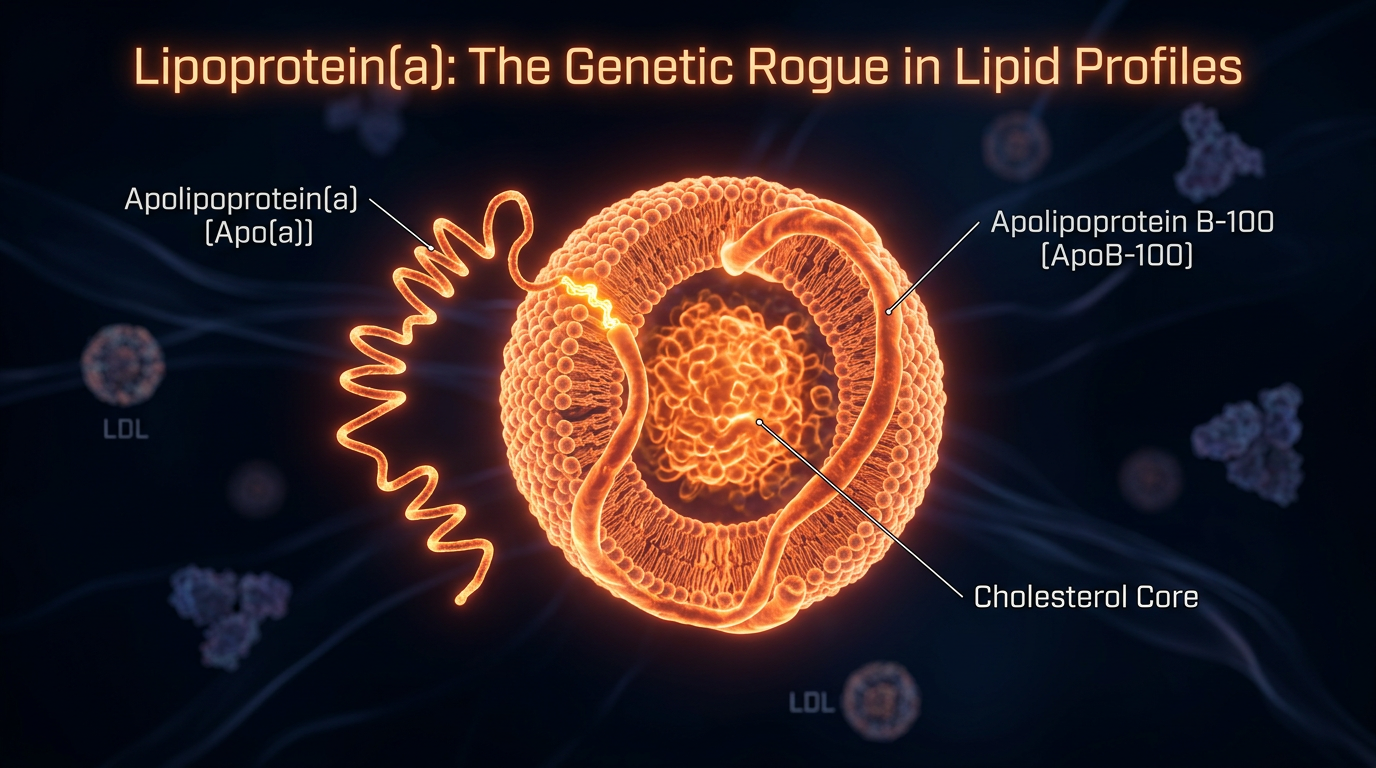
To understand why Lp(a) is so dangerous, one must first understand its unique molecular architecture. At a glance, Lp(a) looks remarkably similar to an LDL particle, but it possesses a critical, lethal modification.
The Anatomy of the Particle
An Lp(a) particle consists of a standard LDL-like core (containing Apolipoprotein B-100 or ApoB), which is covalently bonded to a unique, highly complex protein called Apolipoprotein (a), or apo(a). This bond is formed via a disulphide bridge, linking the two proteins into a single, cohesive unit.
The presence of the apo(a) "tail" transforms a standard cholesterol carrier into a molecular "velcro" that adheres to the arterial walls with terrifying efficiency.
The Kringle Repeats
The structure of the apo(a) protein is defined by "Kringles"—loop-like protein structures named after a Danish pastry because of their shape. Specifically, apo(a) contains Kringle IV and Kringle V domains.
- —The Kringle IV type 2 (KIV-2) segment is polymorphic, meaning the number of repeats varies significantly between individuals.
- —There is an inverse relationship between the number of KIV-2 repeats and the concentration of Lp(a) in the blood.
- —Individuals with fewer repeats produce massive amounts of Lp(a), while those with many repeats produce very little.
The Genetic Blueprint
The production of Lp(a) is governed by the LPA gene located on chromosome 6. Unlike other lipid markers, which are influenced by the environment (the "exposome"), 70% to 90% of the variance in Lp(a) levels is strictly hereditary. This means that for the millions of Britons carrying the high-risk variants, no amount of kale or gym sessions will fundamentally alter the concentration of these particles in their plasma.
---
Mechanisms at the Cellular Level
Lp(a) does not merely "clog" arteries; it orchestrates a multi-front assault on the vascular endothelium through three distinct pathways: Atherogenesis, Thrombosis, and Inflammation.
1. Enhanced Atherogenesis
Standard LDL particles eventually find their way into the arterial wall (the intima) through a process of passive infiltration. However, Lp(a) is actively "sticky." Due to the lysine-binding sites on the apo(a) component, Lp(a) has a high affinity for the extracellular matrix of the arterial wall. It binds to fibrin and glycosaminoglycans, effectively anchoring itself within the vessel wall where it begins to oxidise.
2. Pro-Thrombotic Action (The Plasminogen Mimic)
Perhaps the most dangerous aspect of Lp(a) is its structural similarity to plasminogen, a key enzyme responsible for dissolving blood clots (fibrinolysis).
- —Because apo(a) mimics plasminogen, it competes for the same binding sites on the surface of endothelial cells and fibrin.
- —However, apo(a) cannot be converted into plasmin; it has no clot-dissolving ability.
- —By sitting in the "slot" meant for plasminogen, Lp(a) inhibits the body’s ability to break down clots. This creates a pro-thrombotic state where a small rupture in an arterial plaque leads to a massive, life-ending thrombus (clot).
3. The Carrier of Oxidized Phospholipids (OxPL)
Recent research has revealed that Lp(a) acts as the primary vehicle for Oxidized Phospholipids (OxPL) in the human bloodstream. OxPL are highly reactive molecules that trigger intense inflammatory responses.
- —When Lp(a) delivers OxPL into the arterial wall, it activates macrophages (immune cells).
- —These macrophages ingest the OxPL-laden Lp(a) and transform into foam cells, the foundational building blocks of atherosclerotic plaque.
- —Furthermore, OxPL on Lp(a) stimulates the production of pro-inflammatory cytokines, leading to a state of chronic vascular "simmering."
Critical Mechanism: Lp(a) is essentially a "Triple Threat": it builds the plaque, prevents the body from clearing the plaque's associated clots, and provides the inflammatory fuel to make the plaque unstable.
---
Environmental Threats and Biological Disruptors
While the level of Lp(a) is genetically fixed, the *pathogenicity* of the particle—how much damage it actually does—can be exacerbated by external factors. In the British context, several environmental and biological disruptors act as catalysts for Lp(a)-driven destruction.
Hormonal Shifts
Hormones play a regulatory role in how the liver processes Lp(a).
- —Menopause: It is well-documented that women’s Lp(a) levels often spike during the perimenopausal and postmenopausal transitions. This explains why many women in the UK, who previously had low cardiovascular risk, suddenly experience heart issues in their 50s and 60s.
- —Thyroid Function: Hypothyroidism (an underactive thyroid), which affects a significant portion of the UK population, is known to elevate Lp(a) levels.
The Metabolic Syndrome Overlap
Although Lp(a) is independent of metabolic health, the presence of insulin resistance and high blood sugar (Type 2 Diabetes) creates an environment where Lp(a) is more likely to oxidise. Glycation—the "browning" of proteins due to excess sugar—makes the Lp(a) particle even more "sticky" and inflammatory.
Chronic Inflammation
Inhabitants of modern Britain are often exposed to chronic low-grade inflammation due to ultra-processed diets, sedentary lifestyles, and environmental pollutants. This systemic inflammation upregulates the adhesion molecules on the arterial walls, providing even more "docking stations" for the circulating Lp(a) particles.
---
The Cascade: From Exposure to Disease
The progression of Lp(a)-mediated disease is often referred to as a "cascade" because once the initial damage is done, the biological consequences tumble forward with increasing momentum.
Stage 1: The Initial Deposit
In individuals with high Lp(a), the deposition of particles begins in childhood. Unlike LDL, which often takes decades to form significant plaques, Lp(a) can begin creating fatty streaks in the coronary arteries of teenagers.
Stage 2: Calcification and Stenosis
Lp(a) is uniquely associated with Aortic Stenosis—the narrowing of the heart’s aortic valve. The OxPL carried by Lp(a) promote the deposition of calcium into the valve leaflets. This is a mechanical failure of the heart that often requires open-heart surgery.
Statistic: People in the highest quintile of Lp(a) concentration have a 3-fold increased risk of developing calcific aortic valve stenosis.
Stage 3: The Vulnerable Plaque
Because of its inflammatory cargo, the plaques formed by Lp(a) are often "soft" and "vulnerable." They have a large lipid core and a thin fibrous cap. These are the types of plaques that are prone to sudden rupture.
Stage 4: The Occlusive Event
When a vulnerable plaque ruptures, the pro-thrombotic nature of Lp(a) ensures that the resulting clot is large and difficult to dissolve. This results in a "ST-elevation myocardial infarction" (STEMI)—the most severe form of heart attack—or an ischaemic stroke.
---
What the Mainstream Narrative Omits
The traditional UK medical model, guided largely by NICE (National Institute for Health and Care Excellence), focuses on the QRISK3 algorithm. This tool calculates a person's 10-year risk of a heart attack or stroke based on factors like age, smoking, and *standard* cholesterol.
"The omission is staggering: QRISK3 does not include Lp(a)."
The Statin Paradox
Standard medical advice for high cholesterol is a statin prescription. However, multiple meta-analyses have shown that statins can actually increase Lp(a) levels by 10% to 20%. While statins reduce the overall risk by lowering LDL, for a subset of patients with high Lp(a), the statin might be inadvertently raising their most dangerous lipid marker. This is a truth rarely discussed in GP consultation rooms.
The "Cost-Effectiveness" Barrier
The reason Lp(a) isn't routinely tested in the UK is largely financial. For decades, the argument was: *"Why test for it if we don't have a specific drug to lower it?"* This logic is flawed for several reasons:
- —Risk Stratification: Knowing someone has high Lp(a) allows clinicians to target their *other* risk factors (blood pressure, LDL) much more aggressively.
- —Family Screening: Because it is genetic, one positive test can save the lives of siblings and children through early intervention.
- —Aspirin Therapy: High Lp(a) patients may benefit from low-dose aspirin to counteract the pro-thrombotic effects, a nuance lost when the marker isn't measured.
The Pharmaceutical Pipeline
The mainstream narrative is also slow to adapt because the "cures" for Lp(a) are currently in the hands of high-end biotechnology companies (developing RNA-interference drugs like Pelacarsen and Olpasiran). Until these multi-billion-pound drugs are approved and "pushed" by the industry, the urgency to screen the population remains low.
---
The UK Context
The United Kingdom presents a unique case study in the neglect of Lp(a). The NHS is built on a "population health" model, which prioritises the "average" patient. Unfortunately, the "average" model fails the 20% of the population with high Lp(a).
The Heart UK Initiative
Organisations like HEART UK have been campaigning for universal Lp(a) testing. Currently, the UK consensus is that anyone with premature cardiovascular disease (men under 55, women under 60) or a family history of the same should be tested. However, this is "reactive" rather than "proactive." It waits for the first heart attack to happen before checking the genetics.
The Prevalence in British Ethnicities
The risk is not distributed equally across the UK population.
- —People of South Asian descent, a significant demographic in Britain, tend to have higher levels of Lp(a) and a higher risk of premature heart disease.
- —People of African-Caribbean descent often have the highest absolute levels of Lp(a), though the correlation with heart disease in this group is complex and still being studied.
The GP Knowledge Gap
A significant hurdle in the UK is the knowledge gap among General Practitioners. Many GPs were trained at a time when Lp(a) was considered a "niche" research interest. Consequently, when a patient asks for an Lp(a) test, they are often told it is "unnecessary" or "not supported by local guidelines."
---
Protective Measures and Recovery Protocols
If you discover you have high Lp(a), the mainstream system may offer little hope. However, biological research suggests several avenues for protection and risk mitigation.
1. Aggressive LDL Lowering
Since Lp(a) and LDL both contribute to the total "ApoB" burden (the total number of atherogenic particles), you must lower what you *can* control.
- —PCSK9 Inhibitors: These modern injectables (like Alirocumab or Evolocumab) are available on the NHS for certain high-risk patients. They can lower LDL by 60% and, crucially, can lower Lp(a) by about 20-30%.
- —Ezetimibe: Often used alongside statins to further reduce cholesterol absorption.
2. The Linus Pauling Protocol (Nutraceutical Approach)
Two-time Nobel laureate Linus Pauling proposed a theory that Lp(a) is a "surrogate" for Vitamin C. He argued that when Vitamin C levels are low, the body uses "sticky" Lp(a) to repair the collagen in blood vessels.
- —Protocol: High doses of Vitamin C, L-Lysine, and L-Proline.
- —Mechanism: Lysine and Proline are "binding inhibitors." They occupy the Kringle sites on the Lp(a) molecule, theoretically preventing it from sticking to the arterial wall. While not widely accepted by the NHS, many practitioners use this to successfully reduce vascular inflammation.
3. Niacin (Vitamin B3)
Niacin is one of the few substances known to lower Lp(a) levels by up to 30%. However, it fell out of favour in the UK after the HPS2-THRIVE study, which used a synthetic version of Niacin. Clinical researchers still argue that immediate-release nicotinic acid, when used carefully, is a powerful tool for the Lp(a) patient.
4. Lipoprotein Apheresis
For those with extremely high levels (e.g., >200 nmol/L) and progressive disease, the UK offers Apheresis. This is essentially "dialysis for cholesterol," where the blood is filtered through a machine to physically remove Lp(a) particles. It is highly effective but available only at specialized centres (e.g., in London or Harefield).
5. Managing the "Co-Conspirators"
- —Aspirin: To mitigate the pro-thrombotic risk (under medical supervision).
- —Blood Pressure Control: Keeping pressure low (below 120/80) reduces the "force" pushing Lp(a) into the vessel wall.
- —Anti-Inflammatory Diet: A focus on Omega-3 fatty acids, polyphenols, and the avoidance of seed oils and refined sugars to keep the arterial environment as "cool" as possible.
---
Summary: Key Takeaways
Lipoprotein (a) is the silent driver of the UK’s cardiovascular crisis, hidden in plain sight behind the "LDL" curtain. To protect oneself and one’s family, the following truths must be internalised:
- —It is Genetic: If your parents or siblings had a heart attack before 60, you must assume you are at risk.
- —One Test is Enough: Because it is genetically determined, you only need to test your Lp(a) level once in your lifetime to know your risk profile.
- —Standard Panels are Insufficient: A "normal" cholesterol result does *not* mean you are safe. Demand a specific Lp(a) test (measured in nmol/L for accuracy).
- —Statins are not the Cure: Do not rely on statins alone; they do not lower Lp(a) and may even increase it.
- —Knowledge is the Only Defence: In the absence of a "magic pill" from the NHS, lifestyle optimization, blood pressure management, and specific binding inhibitors (like Lysine) are the primary tools for survival.
The era of ignoring Lp(a) must end. For the millions of Britons carrying this genetic trait, "Innerstanding" this molecule is not just a matter of scientific curiosity—it is a matter of life and death.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Genetic variants associated with increased lipoprotein(a) concentrations are strongly and independently linked to an increased risk of coronary heart disease, suggesting a causal relationship.
Lipoprotein(a) functions as a highly atherogenic and pro-thrombotic particle that significantly contributes to the progression of calcific aortic valve stenosis and coronary artery disease.
International clinical guidelines recommend a one-time measurement of lipoprotein(a) for all adults to identify those at high genetic risk of premature cardiovascular events.
Mendelian randomization studies demonstrate that the magnitude of the effect of lipoprotein(a) on cardiovascular risk is proportional to the absolute change in the mass of circulating particles.
Targeted antisense oligonucleotide therapy significantly reduces lipoprotein(a) levels in patients with cardiovascular disease, providing a potential mechanism for mitigating genetic lipid risk.
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Lp(a): Britain’s Silent Genetic Killer"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper