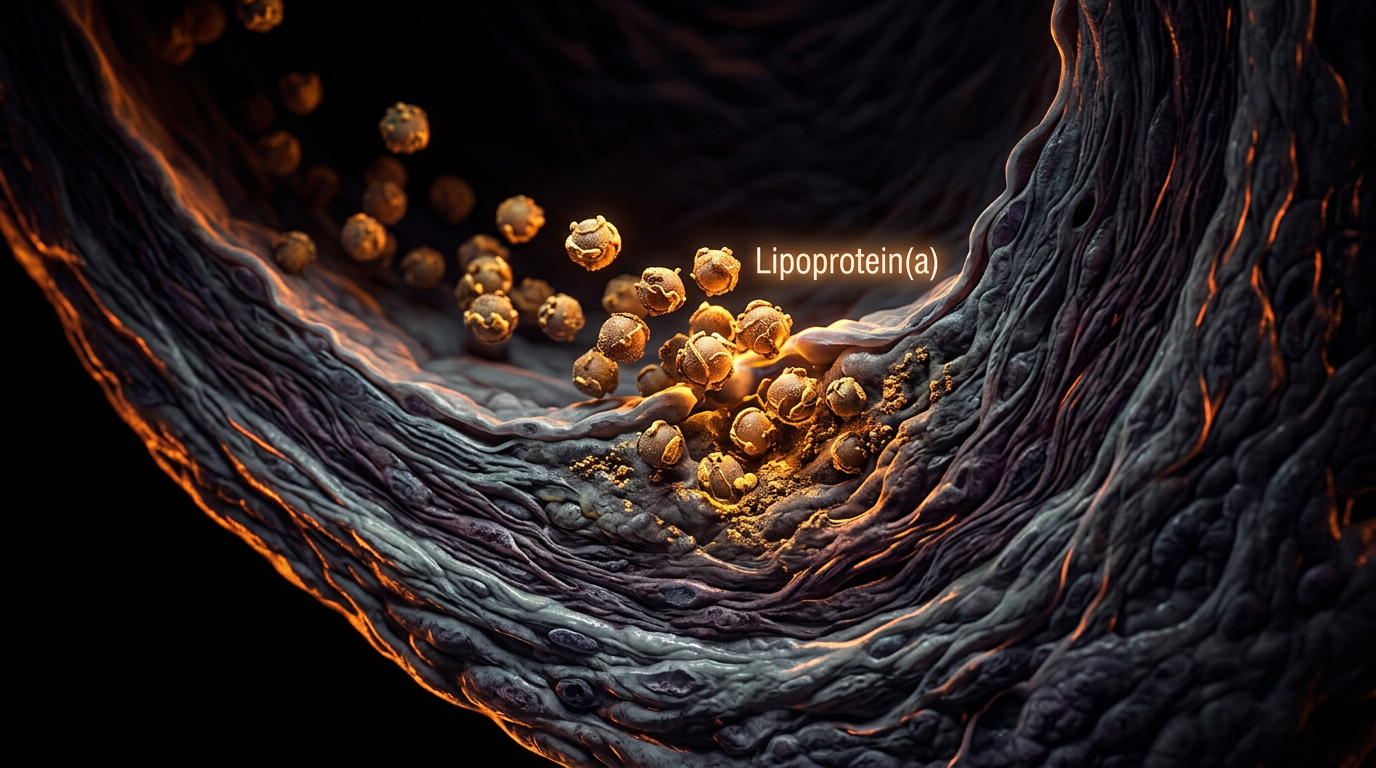
Lipoprotein(a): The Genetic Rogue in Lipid Profiles
Exploring why Lp(a) is a critical independent risk factor for heart disease and the emerging clinical trials aimed at silencing its production.
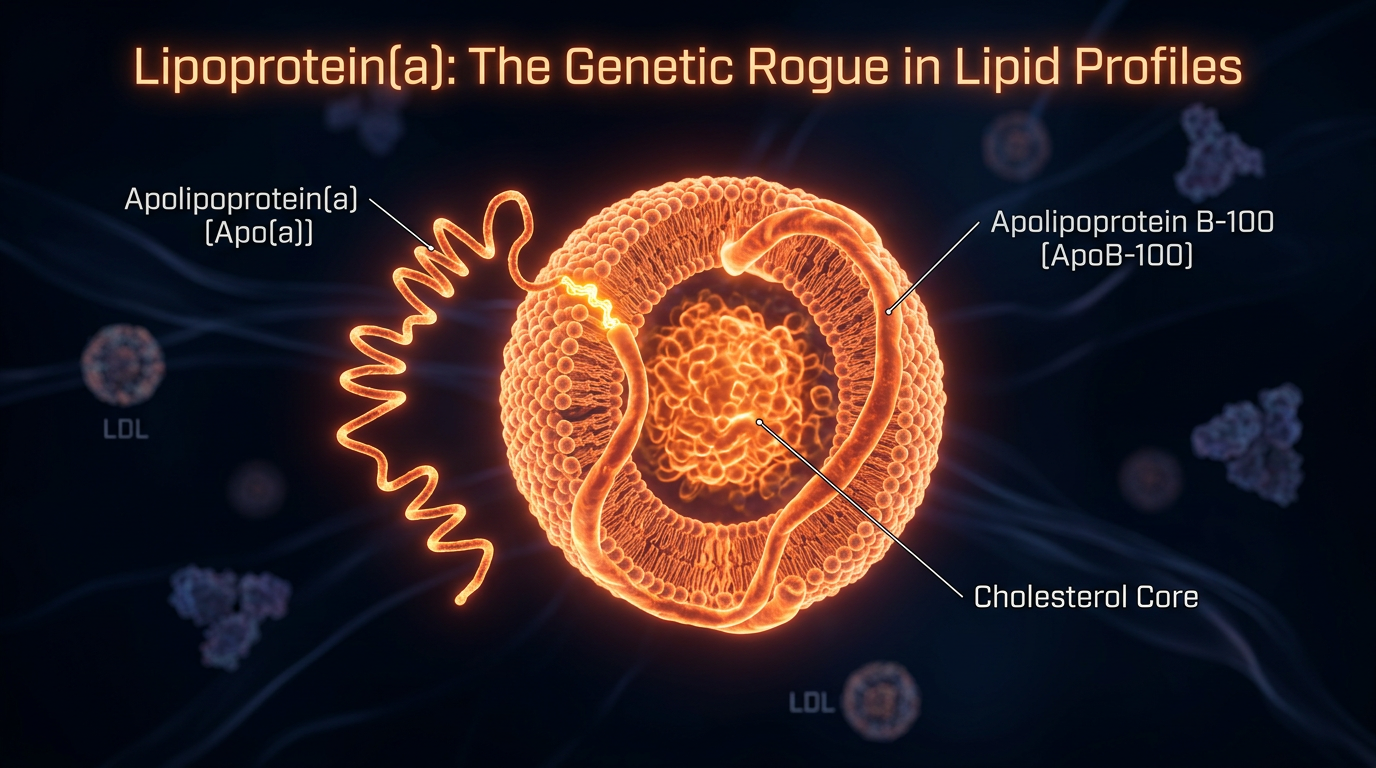
# Lipoprotein(a): The Genetic Rogue in Lipid Profiles
Introduction: The Hidden Architecture of Cardiovascular Risk
In the landscape of modern lipidology, the traditional "cholesterol" narrative is undergoing a radical, albeit overdue, deconstruction. For decades, the medical establishment has focused almost exclusively on Low-Density Lipoprotein (LDL-C)—the so-called ‘bad’ cholesterol—as the primary driver of atherosclerosis. Yet, a significant portion of the population continues to suffer from cardiovascular events despite achieving ‘optimal’ LDL levels. This clinical discrepancy points to a silent, genetically determined saboteur: Lipoprotein(a), or Lp(a).
Known as the ‘genetic rogue,’ Lp(a) is not merely a subset of LDL; it is a distinct, highly inflammatory, and pro-thrombotic particle that remains largely untouched by conventional statin therapy. While the standard lipid panel provides a surface-level overview of metabolic health, the exclusion of Lp(a) testing represents a profound diagnostic blind spot. To achieve true *innerstanding* of one’s cardiovascular destiny, we must look beyond the standard metrics and expose the mechanisms of this molecular interloper.
"In the United Kingdom, it is estimated that approximately 1 in 5 people—roughly 20% of the population—possess elevated levels of Lipoprotein(a) exceeding 50mg/dL (125 nmol/L), significantly increasing their risk of premature heart disease and stroke regardless of lifestyle choices." — *British Heart Foundation / HEART UK Consensus*
---
The Biological Blueprint: Anatomy of a Molecular Mimic
To understand why Lp(a) is so pathological, one must first examine its unique biochemical architecture. Lp(a) consists of an LDL-like particle (containing apolipoprotein B-100) covalently bonded to a specialized protein known as apolipoprotein(a) [apo(a)].
The Apo(a) Appendage
The defining characteristic of Lp(a) is the apo(a) protein, which is synthesised exclusively in the liver. This protein is highly polymorphic, meaning its size varies significantly between individuals based on genetic inheritance. The structural hallmark of apo(a) is the "Kringle" domain—loops of amino acids that resemble a Danish pastry.
Of particular concern is the Kringle IV type 2 (KIV-2) repeat. The fewer KIV-2 repeats an individual inherits, the smaller the apo(a) protein, and paradoxically, the higher the concentration of Lp(a) in the plasma. This inverse relationship is the foundation of the genetic risk: those with "small" Lp(a) particles produce them in much higher quantities.
The Plasminogen Mimicry
The true "rogue" nature of Lp(a) stems from its structural similarity to plasminogen, the enzyme responsible for dissolving blood clots (fibrinolysis). Because apo(a) mimics plasminogen, it competes for binding sites on the vascular wall but lacks the enzymatic activity to break down clots.
- —Pro-thrombotic effect: By inhibiting plasminogen activation, Lp(a) promotes the formation of stable, dangerous clots.
- —Pro-atherogenic effect: Lp(a) binds to the arterial wall more aggressively than standard LDL, delivering its cholesterol payload directly into the intimal layer.
- —Pro-inflammatory effect: Lp(a) is the primary carrier of oxidized phospholipids (OxPL) in the blood. These "rancid" fats trigger a massive immune response, accelerating the calcification of heart valves and the hardening of arteries.
---
The Truth Exposed: Why the "Statin-First" Model Fails
The most uncomfortable truth in contemporary cardiology is that statins—the multi-billion pound "gold standard" for heart health—are largely ineffective against Lp(a). In fact, several high-quality meta-analyses suggest that statins can marginally *increase* Lp(a) levels in certain patients.
The medical establishment’s reluctance to mandate Lp(a) testing stems from a "no drug, no test" philosophy. Since no targeted pharmaceutical "Lp(a)-killer" has been fully commercialised (until the recent advent of RNA-interference therapies), the system has largely ignored the marker. This has left millions of individuals—particularly those with a family history of early myocardial infarction—vulnerable to "residual risk."
"Cardiovascular disease remains the leading cause of death in the UK, accounting for 27% of all deaths. A significant proportion of 'unexplained' heart attacks in physically fit individuals are now attributed to Lp(a) concentrations exceeding the 80th percentile." — *UK Biobank Genetic Study Data*
---
Environmental Disruptors: Epigenetic Pressure on a Genetic Marker
While Lp(a) levels are 70–90% genetically determined by the *LPA* gene, the internal and external environment dictates how "aggressive" these particles become. We must distinguish between the *quantity* of Lp(a) and its *pathogenicity*.
Oxidative Stress and Glycation
The danger of Lp(a) is magnified when it becomes oxidized. In an environment of high oxidative stress—driven by smoking, heavy metal exposure, or chronic systemic inflammation—the phospholipids carried by Lp(a) become toxic. Furthermore, high-sugar diets lead to the glycation of the apo(a) protein, making it more prone to sticking to the arterial walls.
Hormonal Influence
The liver's production of Lp(a) is sensitive to the hormonal milieu. It is well-documented that Lp(a) levels often spike in women during the perimenopause and menopause transition as oestrogen levels decline. This suggests that while the "baseline" is genetic, the "ceiling" can be influenced by the endocrine system.
The Heavy Metal Connection
Emerging research suggests that lead and cadmium exposure can exacerbate the vascular damage caused by Lp(a). These metals promote the endothelial dysfunction that allows Lp(a) to exit the bloodstream and enter the vessel wall, where the process of plaque formation begins.
---
The Diagnostic Gap: Interpreting the Numbers
If you are requesting a lipid panel in the UK, you must specifically ask for "Lipoprotein(a)." A standard "Cholesterol" or "Lipid Profile" does not include it.
What the levels mean:
- —Optimal: < 30 mg/dL (75 nmol/L)
- —Borderline: 30–50 mg/dL (75–125 nmol/L)
- —High Risk: > 50 mg/dL (> 125 nmol/L)
- —Very High Risk: > 180 mg/dL (> 450 nmol/L) — This level is considered a genetic equivalent to Familial Hypercholesterolemia.
*Note: In the UK, labs are moving toward nmol/L (particle concentration) rather than mg/dL (mass), as it is more accurate for smaller, more dangerous particles.*
---
The Recovery Protocol: Mitigating the Rogue Element
Because Lp(a) is resistant to most drugs, recovery and risk mitigation require a sophisticated, multi-targeted approach. We cannot change the *LPA* gene, but we can change the environment in which that gene operates and the stability of the vascular system.
1. The Pauling-Rath Protocol (Vascular Fortification)
Two-time Nobel laureate Linus Pauling and Dr. Matthias Rath proposed that Lp(a) is a "surrogate" for Vitamin C. Their theory suggests that the body produces more Lp(a) to patch up "leaky" blood vessels caused by subclinical scurvy (Vitamin C deficiency).
- —Vitamin C (Ascorbic Acid): 3,000–6,000mg daily. Vitamin C is essential for collagen synthesis, strengthening the basement membrane of arteries.
- —L-Lysine & L-Proline: 2,000–5,000mg daily. These amino acids act as "decoy" binding sites. They bind to the Kringle domains of Lp(a), preventing the particle from attaching to the arterial wall.
2. Niacin (Vitamin B3)
Niacin is one of the few substances known to lower Lp(a) mass by 20–30%. It works by inhibiting the assembly of the Lp(a) particle in the liver.
- —Protocol: High-dose Nicotinic Acid (flushing Niacin) is required, typically 1,000mg to 3,000mg, under medical supervision to monitor liver enzymes.
3. Addressing "Residual Risk" via Inflammation
If your Lp(a) is high, your "fire alarm" is broken. You must ensure there is no "fire" (inflammation) for the Lp(a) to respond to.
- —Lowering hs-CRP: Aim for a High-Sensitivity C-Reactive Protein level of < 1.0 mg/L.
- —Metabolic Flexibility: Maintain HbA1c below 5.3%. Insulin resistance makes Lp(a) significantly more dangerous.
- —Omega-3 Index: Achieve an Omega-3 index of > 8%. High doses of EPA/DHA (3-4g) help reduce the inflammatory potential of the phospholipids attached to Lp(a).
4. Advanced Clinical Interventions
For those in the "Very High Risk" category, natural protocols may need to be supplemented with modern medical advancements:
- —PCSK9 Inhibitors: Injectable drugs like Alirocumab or Evolocumab, while designed for LDL, have been shown to reduce Lp(a) by 25–30%.
- —Lipoprotein Apheresis: A process similar to dialysis where Lp(a) is physically filtered from the blood. This is currently reserved for extreme cases in specialized UK lipid clinics.
- —The Future (ASOs): Antisense Oligonucleotides (like Pelacarsen) are currently in Phase III clinical trials. These "gene-silencing" drugs stop the production of apo(a) at the mRNA level.
---
Lifestyle Strategies for the Lp(a) Patient
Living with high Lp(a) requires a shift in perspective. You are not "sick," but you have a specific vascular vulnerability that demands a high-performance lifestyle.
- —Non-Negotiable Blood Pressure Control: High blood pressure "pushes" Lp(a) into the arterial walls. Aim for 120/80 mmHg or lower.
- —Daily Vascular Shear Stress: Moderate, consistent exercise improves endothelial nitric oxide production, which helps protect the vessel lining from Lp(a) deposition.
- —Avoidance of "Dirty" Fats: Eliminate industrial seed oils (linoleic acid), which easily oxidize and integrate into the Lp(a) particle, increasing its toxicity.
- —Stress Management: Chronic cortisol elevation increases systemic inflammation, acting as a catalyst for Lp(a)-induced plaque rupture.
---
Conclusion: Achieving Innerstanding
Lipoprotein(a) is a relic of our evolutionary past—likely developed to protect our ancestors from excessive bleeding or to compensate for the loss of endogenous Vitamin C production. In the modern world of processed foods, sedentary lifestyles, and environmental toxins, this protective mechanism has become a "genetic rogue."
To ignore Lp(a) is to gamble with one's cardiovascular future. High-authority lipid management requires a departure from the "statin-for-all" dogma and a move toward precision medicine. By identifying this marker early, fortifying the vascular wall through the Pauling-Rath protocol, and ruthlessly eliminating systemic inflammation, the risk posed by this rogue particle can be effectively neutralised.
True health is not the absence of genetic risk, but the mastery of the environment that governs it. Take the test, know your numbers, and secure the foundation of your biological architecture.
*
"References & Further Reading:"
- —*The HEART UK Consensus Statement on Lipoprotein(a).*
- —*The Lancet: Lipoprotein(a) as a Genetic Risk Factor for Coronary Artery Disease.*
- —*Journal of the American College of Cardiology: Lp(a) and its role in Calcific Aortic Valve Stenosis.*
- —*The Pauling-Rath Unified Theory of Human Cardiovascular Disease.*
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
High levels of lipoprotein(a) are genetically determined and represent an independent, causal risk factor for cardiovascular disease and aortic valve stenosis.
Genetic variants at the LPA locus are more strongly associated with the risk of coronary artery disease than any other lipid-related genetic markers.
Targeted antisense oligonucleotide therapy can effectively lower lipoprotein(a) concentrations in individuals with elevated levels, addressing a previously untreatable genetic risk factor.
Lipoprotein(a) promotes atherosclerosis and thrombosis through its unique structure, which includes an LDL-like particle and the highly polymorphic apolipoprotein(a).
Mendelian randomization studies suggest that large absolute reductions in lipoprotein(a) are necessary to achieve clinically meaningful decreases in cardiovascular event risk.
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Lipoprotein(a): The Genetic Rogue in Lipid Profiles"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper