Lipoprotein(a): The Genetic 'Hidden' Heart Risk Missing from Standard NHS Blood Panels
Explore why this inherited lipid variant might be the true driver of premature heart attacks despite normal LDL levels.
Overview
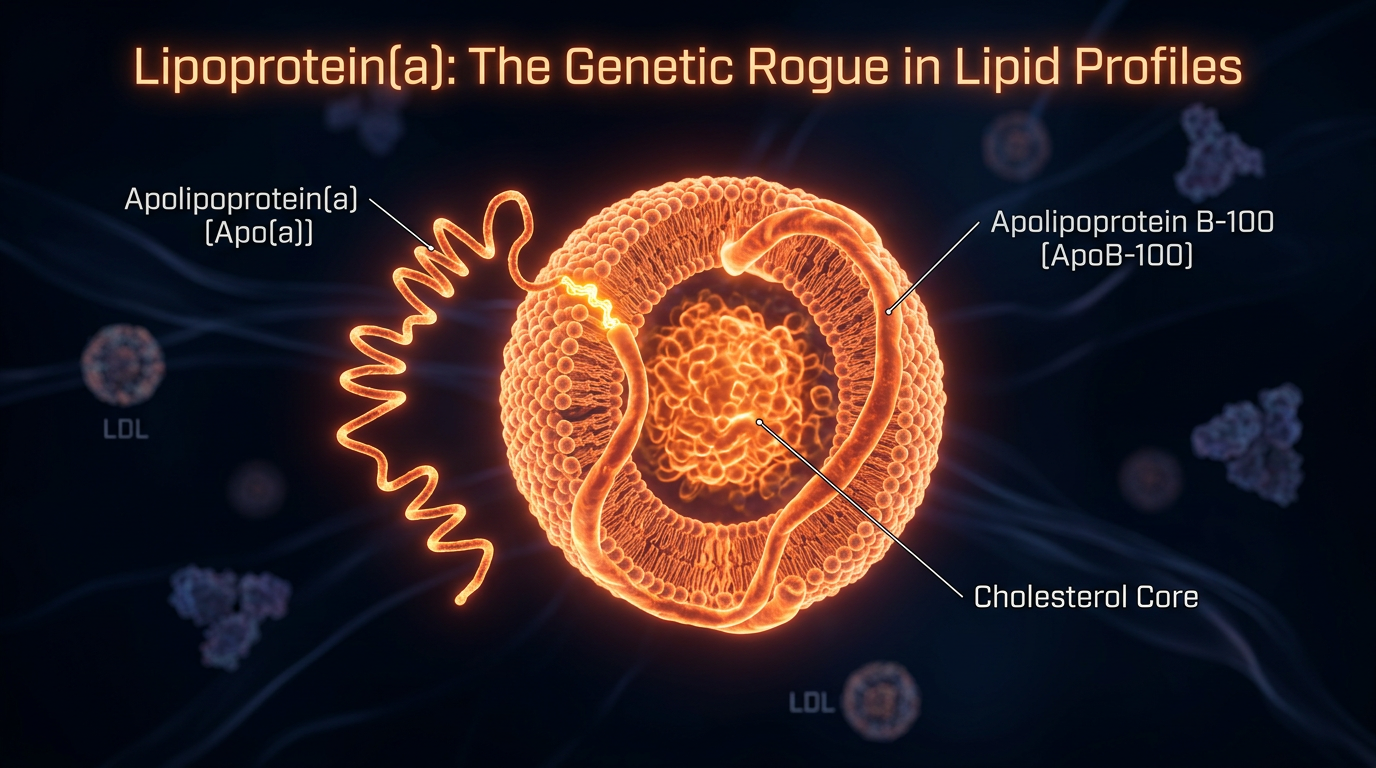
Lipoprotein(a), hereafter referred to as Lp(a), represents perhaps the most significant clandestine driver of premature atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valve stenosis (CAVS) within the British population. Despite its potent pathogenicity, it remains a systemic oversight in standard NHS primary care lipid panels, which typically prioritise the quantification of low-density lipoprotein cholesterol (LDL-C) and high-density lipoproteins (HDL). Structurally, Lp(a) is a sophisticated macromolecular complex consisting of an LDL-like particle, in which a central core of cholesteryl esters is enveloped by apolipoprotein B-100 (ApoB). However, its unique and devastating virulence is derived from the covalent attachment of a polymorphic glycoprotein, apolipoprotein(a) [Apo(a)], via a single disulphide bridge. This biochemical architecture transforms a standard lipid carrier into a multi-pronged instrument of vascular damage, operating through three distinct, synergistic pathways: pro-atherogenic, pro-thrombotic, and pro-inflammatory.
The concentration of Lp(a) in the plasma is determined almost exclusively—upwards of 90%—by variation at the *LPA* gene locus on chromosome 6q25.6. Unlike conventional LDL-C, which is highly sensitive to nutritional intervention and metabolic flux, Lp(a) levels remain remarkably stable throughout an individual’s life, unaffected by statins, exercise, or dietary modification. This genetic determinism, characterised by the inverse relationship between the number of Kringle IV type 2 (KIV-2) repeats and circulating mass, means that approximately 20% of the UK population harbours elevated levels (above 125 nmol/L or 50 mg/dL) that place them at an exponentially higher risk of myocardial infarction and stroke, often despite seemingly "healthy" standard cholesterol readings.
The biological mechanism of Lp(a) is a masterclass in evolutionary mimicry. Due to the high structural homology between Apo(a) and plasminogen, Lp(a) competitively inhibits the activation of plasmin, thereby impairing fibrinolysis. This creates a pro-thrombotic environment where blood clots are less likely to be dissolved, significantly increasing the lethality of plaque rupture. Furthermore, as highlighted in research published in *The Lancet* and *Nature Reviews Cardiology*, Lp(a) serves as the primary plasma carrier for oxidised phospholipids (OxPL). These OxPL molecules trigger a cascade of pro-inflammatory cytokines within the arterial intima, accelerating the recruitment of macrophages and the formation of foam cells. At INNERSTANDIN, we recognise that the failure to routinely screen for this genetic variant represents a critical diagnostic gap. By neglecting the *LPA* genotype, the current clinical paradigm overlooks a fundamental driver of "residual risk," leaving millions of individuals vulnerable to vascular events that are entirely predictable through advanced proteomic and genomic analysis. The evidence is unequivocal: Lp(a) is not merely a lipid sub-fraction, but an independent, causal risk factor that necessitates a radical shift in how we approach cardiovascular preventative medicine.
The Biology — How It Works
To grasp why Lipoprotein(a) [Lp(a)] represents a physiological "silent assassin," one must look beyond the crude lipid metrics typically utilised in NHS primary care. At a molecular level, Lp(a) is an evolutionary enigma: a low-density lipoprotein (LDL)-like particle covalently bonded to a unique, highly polymorphic glycoprotein known as apolipoprotein(a) [apo(a)]. This linkage occurs via a single disulphide bridge between the apolipoprotein B-100 (apoB) component and the apo(a) chain. While standard lipid panels measure the cholesterol content within LDL, they remain blind to the presence of apo(a), a structural addition that transforms a standard lipid transporter into a potent mediator of vascular destruction.
The pathogenicity of Lp(a) is driven by three distinct biological mechanisms: pro-atherogenesis, pro-thrombosis, and pro-inflammation. Unlike LDL-C, which fluctuates based on dietary and lifestyle interventions, the plasma concentration of Lp(a) is 80–90% genetically determined by the *LPA* gene locus on chromosome 6q25.3. Within this locus, the number of "Kringle IV type 2" (KIV-2) repeat sequences dictates the size of the apo(a) isoform. Crucially, there is an inverse correlation between the number of KIV-2 repeats and plasma Lp(a) concentrations; individuals with fewer repeats synthesise smaller, high-velocity isoforms that reach pathological levels. This genetic determinism is why INNERSTANDIN emphasises that standard "healthy" lifestyle markers often mask an underlying, inherited cardiovascular vulnerability.
The pro-thrombotic nature of Lp(a) arises from the striking structural homology between apo(a) and plasminogen. Because apo(a) mimics the Kringle domains of plasminogen but lacks its catalytic protease activity, it acts as a competitive antagonist. It binds to fibrin and endothelial cell receptors, effectively blocking the conversion of plasminogen to plasmin. This inhibition of fibrinolysis ensures that micro-thrombi, which might otherwise be dissolved by the body’s innate haemostatic mechanisms, persist and stabilise within the arterial wall. Evidence published in *The Lancet* and *Journal of the American College of Cardiology* confirms that this "molecular mimicry" is a primary driver of myocardial infarction in patients with otherwise unremarkable cholesterol profiles.
Furthermore, Lp(a) serves as the primary plasma vehicle for oxidised phospholipids (OxPL). These OxPLs are highly reactive and trigger a cascade of innate immune responses upon entering the sub-endothelial space. Macrophages that ingest Lp(a) become foam cells more rapidly than those ingesting LDL, while simultaneously releasing pro-inflammatory cytokines such as IL-6 and TNF-α. In the context of the aortic valve, the delivery of OxPL by Lp(a) promotes the transition of valvular interstitial cells into an osteoblastic phenotype, leading to rapid calcification. This is not merely a "lipid problem"; it is a complex, multi-systemic assault on the vascular and valvular architecture that the current NHS "Statins-for-all" paradigm fails to address. For those seeking true biological INNERSTANDIN, recognising the dual role of Lp(a) as both a cholesterol carrier and a thrombogenic blockade is essential for survival.
Mechanisms at the Cellular Level
To comprehend why Lipoprotein(a) [Lp(a)] represents an insidious departure from the traditional lipid paradigm, one must interrogate its unique molecular architecture. Structurally, Lp(a) consists of a low-density lipoprotein (LDL)-like particle, wherein a single molecule of apolipoprotein B-100 (apoB) is covalently tethered to the highly polymorphic glycoprotein, apolipoprotein(a) [apo(a)], via a single disulphide bridge. At INNERSTANDIN, we recognise that this structural appendage transforms a standard lipid carrier into a potent, multi-modal pathological agent. Unlike LDL-C, which is primarily a metabolic byproduct of VLDL remodelling, Lp(a) levels are nearly entirely genetically determined by the *LPA* gene, remaining recalcitrant to standard NHS interventions like statins or lifestyle modifications.
The cellular pathogenicity of Lp(a) is driven by a trifecta of mechanisms: it is simultaneously pro-atherogenic, pro-thrombotic, and pro-inflammatory. Firstly, the particle’s atherogenicity is amplified by its heightened affinity for the subendothelial extracellular matrix. Once Lp(a) infiltrates the tunica intima, the apo(a) component binds aggressively to proteoglycans and fibronectin. This sequestering within the arterial wall facilitates the oxidation of the particle, a process accelerated by the fact that Lp(a) is the primary plasma carrier of oxidised phospholipids (OxPL). Peer-reviewed research, notably in *The Lancet*, suggests that these OxPLs trigger a robust inflammatory response by activating toll-like receptors on macrophages, inducing a phenotype shift toward highly unstable, cytokine-secreting foam cells.
Secondly, the "molecular mimicry" of apo(a) poses a direct threat to the fibrinolytic system. Apo(a) shares a high degree of structural homology with plasminogen, specifically containing repeated sequences known as Kringle domains (specifically KIV and KV). Because of this resemblance, Lp(a) competitively inhibits plasminogen binding to fibrin and the endothelial surface. By displacing plasminogen, Lp(a) suppresses the generation of plasmin, effectively stalling the dissolution of clots. This creates a pro-thrombotic environment where minor endothelial erosions, which might otherwise be resolved, escalate into occlusive events such as myocardial infarction or ischaemic stroke.
Furthermore, the impact extends to the aortic valve. Research indexed in PubMed highlights that Lp(a)-associated OxPLs are internalised by valvular interstitial cells, promoting an osteogenic gene programme. This leads to the deposition of hydroxyapatite, driving calcific aortic valve stenosis—a condition for which there is currently no pharmacological treatment in the UK. By bypassing the standard LDL receptors and persisting in the circulation for extended half-lives, Lp(a) acts as a persistent metabolic toxin. For the INNERSTANDIN community, it is clear: the failure to include Lp(a) in routine NHS lipid panels is a critical oversight, as it ignores a particle that is qualitatively more dangerous than the LDL-C targets currently prioritised by clinical guidelines.
Environmental Threats and Biological Disruptors
While the serum concentration of Lipoprotein(a) [Lp(a)] is approximately 90% genetically predetermined via the LPA locus, its pathological virulence is not a static variable; rather, it is significantly modulated by the systemic environment and external biological disruptors. At INNERSTANDIN, we recognise that the structural complexity of Lp(a)—specifically the covalent linkage of apolipoprotein(a) to an LDL-like particle via a disulphide bridge—renders it uniquely susceptible to oxidative modification. In the modern UK landscape, environmental stressors act as catalytic "second hits" that transform a genetically high Lp(a) profile from a latent risk into an active vascular catastrophe.
One of the primary biological disruptors influencing Lp(a) pathogenicity is the presence of oxidised phospholipids (OxPL). Research published in *The Lancet* and the *Journal of the American College of Cardiology* demonstrates that Lp(a) is the primary carrier of OxPL in human plasma. Environmental pollutants, particularly particulate matter (PM2.5) prevalent in urban UK centres, induce systemic oxidative stress that facilitates the oxidation of these phospholipids. Once oxidised, the OxPL-Lp(a) complex triggers a potent pro-inflammatory cascade within the endothelium, mediated by the activation of macrophages and the subsequent release of IL-1β and TNF-α. This environmental priming of the Lp(a) particle accelerates the transition from simple cholesterol deposition to complex, unstable atherosclerotic plaques.
Furthermore, the structural homology between the kringle domains of apo(a) and plasminogen creates a state of competitive inhibition that is exacerbated by environmental disruption of the fibrinolytic system. In a biological milieu characterised by chronic systemic inflammation—often driven by the "Western" dietary pattern or circadian rhythm disruption—the body’s innate ability to dissolve micro-thrombi is already compromised. Lp(a) disrupts the binding of plasminogen to the endothelial surface, effectively paralysing endogenous thrombolysis. When this genetic predisposition meets environmental disruptors like heavy metal exposure (e.g., lead or cadmium) or chronic psychosocial stress, the vascular endothelium loses its hemostatic balance, leading to the "hidden" myocardial infarctions often seen in otherwise "low-cholesterol" NHS patients.
Biological disruptors also include the specific hormonal shifts often overlooked in standard clinical pathways. Evidence suggests that the decline in oestrogen during menopause or the use of certain endocrine-disrupting chemicals (EDCs) can lead to a significant upward shift in Lp(a) expression. This epigenetic-environmental interplay suggests that the "genetic fixity" of Lp(a) is a reductive myth. At INNERSTANDIN, we emphasise that while the NHS blood panels remain fixated on LDL-C, they ignore how environmental toxins and metabolic dysfunction turn Lp(a) into a pro-thrombotic and pro-inflammatory weapon. The internal biological environment determines whether an elevated Lp(a) level remains a statistical quirk or becomes a terminal event. Understanding these disruptors is paramount for moving beyond the archaic lipid hypothesis into a more nuanced, evidence-led paradigm of cardiovascular proteostasis.
The Cascade: From Exposure to Disease
The pathogenicity of Lipoprotein(a) [Lp(a)] is not merely a quantitative amplification of low-density lipoprotein (LDL) risk; it represents a distinct, multi-modal assault on vascular integrity that standard lipid profiling systematically ignores. To achieve a true INNERSTANDIN of this silent driver of atherosclerotic cardiovascular disease (ASCVD), one must dissect the tripartite mechanism of its cascade: pro-atherogenic, pro-thrombotic, and pro-inflammatory.
Structurally, Lp(a) consists of an LDL-like particle in which a molecule of apolipoprotein B-100 (apoB) is covalently linked to the highly polymorphic glycoprotein, apolipoprotein(a) [apo(a)], via a singular disulphide bridge. This complex is inherently more hazardous than LDL. Due to its unique protein composition, Lp(a) exhibits an increased affinity for the arterial wall’s subendothelial proteoglycans. Once sequestered within the intima, the apo(a) component facilitates the entrapment of the entire particle, leading to the accelerated formation of the fatty streak. Unlike standard LDL, which requires significant oxidation to become pathogenic, Lp(a) arrives pre-loaded with a cargo of oxidised phospholipids (OxPL). These OxPLs are not passive observers; they are potent bioactive molecules that trigger the expression of adhesion molecules such as VCAM-1 and ICAM-1 on endothelial cells, stimulating the recruitment of circulating monocytes and their subsequent transformation into foam cells.
The most insidious element of the Lp(a) cascade—and perhaps the most overlooked in UK clinical practice—is its structural homology with plasminogen. The apo(a) molecule contains multiple 'Kringle' domains, specifically Kringle IV type 2 (KIV-2), which mimics the fibrin-binding domains of plasminogen. This molecular mimicry creates a state of competitive inhibition; Lp(a) binds to fibrin and endothelial cell receptors, preventing plasminogen from being converted into plasmin. The result is a profound suppression of fibrinolysis. In the event of a plaque rupture, this pro-thrombotic environment ensures that a thrombus is not effectively dissolved, significantly increasing the risk of acute myocardial infarction or ischaemic stroke. Evidence published in *The Lancet* and the *European Heart Journal* confirms that this "plasminogen interference" places individuals with high Lp(a) at a risk level that LDL-C metrics cannot account for.
Furthermore, the INNERSTANDIN of the systemic impact extends to the aortic valve. The OxPLs carried by Lp(a) are primary drivers of calcific aortic valve stenosis (CAVS). Through the action of autotaxin—an enzyme that converts lysophosphatidylcholine into lysophosphatidic acid—Lp(a) promotes the osteogenic differentiation of valvular interstitial cells. This leads to the progressive deposition of hydroxyapatite, rigidifying the valve and necessitating surgical intervention. Given that Lp(a) levels are 70–90% genetically determined by the *LPA* gene locus and remain largely unaffected by the statin therapies standard to the NHS, the failure to screen for this particle represents a significant diagnostic gap in British preventative cardiology. The cascade from exposure to disease is a lifelong accumulation of vascular and valvular insult, unmitigated by conventional lifestyle or pharmaceutical interventions.
What the Mainstream Narrative Omits
The standard lipid panel prioritised by the NHS—focusing almost exclusively on total cholesterol, high-density lipoprotein (HDL), and low-density lipoprotein cholesterol (LDL-C)—remains fundamentally incomplete, leaving a critical diagnostic void regarding the most potent genetically determined risk factor for myocardial infarction and stroke: Lipoprotein(a) [Lp(a)]. While the mainstream narrative operates under the reductive assumption that lowering LDL-C is the panacea for cardiovascular risk reduction, it systematically overlooks the idiosyncratic pathogenicity of the Lp(a) particle, which is chemically distinct and physiologically more aggressive than standard LDL.
At the molecular level, Lp(a) consists of an LDL-like particle containing apolipoprotein B-100 (apoB-100) covalently linked via a single disulphide bridge to a highly polymorphic glycoprotein: apolipoprotein(a) [apo(a)]. The presence of apo(a) is what renders this particle particularly deleterious. Research published in *The Lancet* and the *European Heart Journal* highlights a dual mechanism of harm: pro-atherogenic and pro-thrombotic. The structural homology between apo(a) and plasminogen is the primary biological "glitch" omitted from public health discourse. Because apo(a) mimics the Kringle domains of plasminogen, it competitively inhibits the binding of plasminogen to fibrin and the surface of endothelial cells. This blockade prevents the activation of plasmin, effectively crippling the body’s endogenous fibrinolytic system and promoting a pro-thrombotic environment that accelerates the formation of recalcitrant clots within the coronary and carotid arteries.
Furthermore, Lp(a) acts as the primary vehicle for oxidised phospholipids (OxPL) in the human bloodstream. These OxPLs are not mere metabolic byproducts; they are potent triggers for the nuclear factor-kappa B (NF-κB) signalling pathway within the vascular endothelium. This induces the expression of adhesion molecules and the recruitment of monocyte-derived macrophages, transforming the arterial wall into a site of chronic, non-resolving inflammation. Unlike LDL-C, which is highly sensitive to dietary intervention and aerobic exercise, circulating levels of Lp(a) are 70–90% genetically determined by the *LPA* locus on chromosome 6q25-27. Specifically, the Kringle IV type 2 (KIV-2) copy number variation dictates the size of the apo(a) isoforms; individuals with fewer KIV-2 repeats synthesise smaller, more concentrated, and highly atherogenic particles.
The failure of standard UK blood panels to assess Lp(a) means that approximately 20% of the population—millions of individuals—harbour a "hidden" risk that remains invisible to current NICE-guided screening protocols. At INNERSTANDIN, we recognise that relying on LDL-C alone is akin to assessing a structural fire while ignoring the presence of chemical accelerants. Until Lp(a) quantification becomes a standardised component of the cardiovascular risk profile, the medical establishment is effectively ignoring a mendelian driver of premature death that cannot be "statined" or "dieted" away, but requires targeted antisense oligonucleotide or siRNA therapies to silence the *LPA* gene expression at its source.
The UK Context
Within the current landscape of British clinical practice, a systemic diagnostic void persists regarding the primary prevention of atherosclerotic cardiovascular disease (ASCVD). The standard NHS lipid profile—comprising Total Cholesterol, HDL-C, and calculated LDL-C—operates on an obsolete paradigm that ignores the pathogenic potency of Lipoprotein(a) [Lp(a)]. This genetically determined particle, present in high concentrations in approximately 20% of the UK population, remains invisible to routine screening until a premature coronary event necessitates tertiary investigation. At INNERSTANDIN, we recognise this as a critical failure in genomic-led medicine.
The biological hazard of Lp(a) is derived from its unique molecular architecture: a low-density lipoprotein-like particle covalently bonded to apolipoprotein(a) [apo(a)] via a disulphide bridge. This apo(a) moiety possesses a striking structural homology with plasminogen, containing "kringle" domains that facilitate competitive inhibition of fibrinolysis. Research published in *The Lancet* and data extracted from the UK Biobank—a cohort of half a million UK participants—confirm that Lp(a) acts as a dual-threat agent: it is both highly pro-atherogenic and pro-thrombotic. Unlike standard LDL-C, which can be modulated through dietary intervention, Lp(a) levels are 70–90% genetically determined by the *LPA* locus on chromosome 6q25.2-q27. Consequently, millions of UK citizens maintaining "healthy" lifestyle markers remain at high risk for myocardial infarction and calcific aortic valve stenosis (CAVS) due to inherited elevations in Lp(a) that traditional statin therapies fail to attenuate.
The disparity between academic consensus and NHS implementation is stark. While HEART UK and the European Society of Cardiology (ESC) recommend that every adult should have their Lp(a) measured at least once in their lifetime, NICE (National Institute for Health and Care Excellence) guidelines currently limit testing to those with a family history of premature CVD or familial hypercholesterolaemia. This conservative stance ignores the "residual risk" prevalent in the UK population, where individuals with optimally managed LDL-C continue to experience vascular events. By failing to integrate Lp(a) quantification—specifically using isoform-independent assays expressed in nmol/L—into standard blood panels, the UK healthcare system remains reactive rather than proactive. INNERSTANDIN advocates for the immediate recognition of Lp(a) as a primary causal risk factor, essential for shifting the UK's cardiovascular strategy toward true precision biological management.
Protective Measures and Recovery Protocols
Mitigating the pathological footprint of Lipoprotein(a) [Lp(a)] requires a departure from traditional lipid-lowering paradigms, as the plasma concentration of this highly atherogenic particle is approximately 70–90% genetically determined by the *LPA* locus on chromosome 6. Because Lp(a) remains largely recalcitrant to standard HMG-CoA reductase inhibitors (statins)—which may paradoxically increase Lp(a) levels by up to 20%—recovery and protection protocols must focus on radical LDL-C suppression, interference with the plasminogen-mimetic mechanism, and the emerging frontier of RNA therapeutics.
The primary biological objective in high-Lp(a) phenotypes is the systemic reduction of the overall apolipoprotein B (ApoB) burden to offset the independent risk of the Lp(a) particle. Evidence from the FOURIER and ODYSSEY OUTCOMES trials suggests that PCSK9 inhibitors, such as evolocumab and alirocumab, offer a dual-pathway benefit. They not only lower LDL-C to physiologic nadirs but also facilitate a 20–30% reduction in Lp(a) by increasing the availability of hepatic LDL receptors and potentially enhancing the clearance of Lp(a) via the plasminogen receptor. For patients within the UK clinical context who fall outside the strict NICE criteria for these biologics, the INNERSTANDIN approach necessitates a rigorous focus on endothelial integrity to prevent the sequestration of the Lp(a) particle within the sub-endothelial space.
The "Linus Pauling" protocol, though often marginalised in conventional NHS primary care, offers a compelling biochemical rationale for recovery. Lp(a) acts as a "molecular patch" for weakened vascular walls, binding to lysine residues in damaged collagen. By saturating the system with exogenous L-Lysine and L-Proline, clinicians can theoretically inhibit the binding of the apo(a) kringle domains to the vascular endothelium, promoting the detachment of deposited particles. When coupled with high-dose Ascorbate (Vitamin C) to stimulate collagen synthesis, this protocol addresses the structural vulnerability that makes Lp(a) so lethal.
Furthermore, the pro-thrombotic nature of Lp(a)—driven by its structural homology to plasminogen—requires a targeted anti-platelet strategy. Because the apo(a) component competitively inhibits plasminogen activation, individuals with elevated levels exist in a state of suppressed fibrinolysis. Recovery protocols should consider low-dose aspirin or natural fibrinolytic agents like Nattokinase to counteract this "sticky blood" phenomenon.
In refractory cases, Lipoprotein Apheresis remains the most potent, albeit invasive, protective measure available in specialised UK lipid centres. This extracorporeal process physically removes Lp(a) and its associated oxidised phospholipids (OxPL) from the circulation. However, the future of INNERSTANDIN-grade cardiovascular recovery lies in antisense oligonucleotides (ASOs) and small interfering RNA (siRNA) therapies. Agents like Pelacarsen and Olpasiran, currently in Phase III trials, target the *LPA* mRNA in the liver, achieving sustained reductions in Lp(a) of up to 90%. Until these become standard of care, the priority must be aggressive global risk factor modification and the stabilisation of the endothelium to neutralise the genetic "hidden" threat that standard NHS panels systematically ignore.
Summary: Key Takeaways
Lipoprotein(a) [Lp(a)] represents a distinct, genetically determined lipid fraction that traditional NHS lipid profiles fundamentally fail to quantify, leaving millions of patients in the UK under-stratified for cardiovascular risk. Structurally, the moiety consists of a low-density lipoprotein-like particle wherein apolipoprotein B-100 is covalently linked via a disulphide bridge to the highly polymorphic apolipoprotein(a). This unique architecture facilitates a pathogenic triad: pro-atherogenic cholesterol delivery, pro-inflammatory oxidation via sequestered oxidised phospholipids (OxPL), and pro-thrombotic activity due to the protean structural homology between apo(a) and plasminogen. At INNERSTANDIN, we recognise that the competitive inhibition of plasminogen activation by Lp(a) induces a state of hypofibrinolysis, significantly elevating the risk of myocardial infarction and ischaemic stroke independent of standard LDL-C levels.
Evidence from Mendelian randomisation studies and large-scale meta-analyses published in *The Lancet* underscores that elevated Lp(a) is a causal driver of calcific aortic valve stenosis (CAVS), a condition frequently misattributed solely to age-related degeneration. Because circulating Lp(a) concentrations are approximately 90% genetically predetermined by the *LPA* locus, standard lifestyle interventions and statin therapies remain largely ineffective at reducing its concentration, necessitating a shift toward emerging antisense oligonucleotide and RNA interference technologies. The current clinical omission of universal Lp(a) screening in the UK represents a profound diagnostic lacuna, masking a potent, heritable driver of premature vascular senescence and systemic endothelial dysfunction. To achieve true biological INNERSTANDIN, one must look beyond the standard panel to the underlying genetic architecture of the lipidome.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Elevated lipoprotein(a) is an independent, genetic, and causal risk factor for cardiovascular disease that is not captured by standard low-density lipoprotein cholesterol measurements.
Consensus statements emphasize that lipoprotein(a) concentrations are approximately 90% genetically determined and remain largely unaffected by lifestyle changes or standard statin therapy.
Lipoprotein(a) contributes to cardiovascular risk through its pro-atherogenic, pro-inflammatory, and potentially pro-thrombotic properties mediated by its apolipoprotein(a) component.
Meta-analysis data indicates that individuals with lipoprotein(a) levels above 50 mg/dL face a significantly higher risk of myocardial infarction and stroke regardless of their LDL cholesterol status.
Targeted antisense oligonucleotides have demonstrated the ability to reduce lipoprotein(a) levels by up to 80%, addressing a major gap in current lipid-lowering pharmacological interventions.
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Lipoprotein(a): The Genetic 'Hidden' Heart Risk Missing from Standard NHS Blood Panels"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Cardiovascular Health — products curated by our research team for educational relevance and biological support.

Magnesium Blend – The Most Important Mineral

Clean Slate – Detoxes thousands of chemicals,heavy metals, pesticides, allergens, mold spores and fungus

Vegan Essential Amino Acids – Plant-Powered Protein Building
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper