Metabolic Dyshomeostasis: The Link Between Iron Overload and Beta-Cell Oxidative Stress
Analyze the mechanistic link between iron sequestration and beta-cell oxidative stress, detailing how mineral imbalances disrupt insulin signaling and metabolic regulation.

Overview
The prevailing clinical narrative surrounding Type 2 Diabetes Mellitus (T2DM) has historically focused on glucotoxicity and lipotoxicity as the dual engines of metabolic decline. However, at INNERSTANDIN, we move beyond these superficial markers to expose the more insidious role of iron-mediated oxidative stress in the destruction of pancreatic architecture. Metabolic dyshomeostasis, particularly in the context of iron overload, represents a critical yet under-appreciated intersection of bioinorganic chemistry and endocrinology. While iron is an essential cofactor for mitochondrial respiration and oxygen transport, its chemical volatility makes it a potent catalyst for cellular demise when systemic sequestration mechanisms fail.
The "iron-insulin axis" describes a bidirectional pathological relationship where excess iron drives insulin resistance, and hyperinsulinaemia further facilitates iron accumulation within metabolic tissues. Central to this failure is the pancreatic beta-cell, a unit of high metabolic demand but strikingly low antioxidant defences. Unlike hepatocytes or myocytes, beta-cells exhibit disproportionately low expressions of protective enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase. Consequently, when the Labile Iron Pool (LIP) expands due to hereditary haemochromatosis, chronic dietary surfeit, or repeated transfusions, the beta-cell is subjected to relentless oxidative assault. Through the Fenton and Haber-Weiss reactions, free divalent iron ($Fe^{2+}$) facilitates the conversion of hydrogen peroxide into the highly reactive hydroxyl radical ($OH^\bullet$), leading to lipid peroxidation of the plasma membrane, protein carbonylation, and DNA fragmentation.
Evidence derived from the UK Biobank and longitudinal studies published in *The Lancet Diabetes & Endocrinology* suggests that even high-normal ferritin levels—well below the threshold for clinical haemochromatosis—correlate significantly with an increased risk of glucose intolerance. This subclinical iron overload acts as a "silent" driver of beta-cell ferroptosis, a form of regulated cell death distinct from apoptosis, characterised by iron-dependent lipid peroxidation. Furthermore, excess iron impairs the glucose-stimulated insulin secretion (GSIS) mechanism by disrupting mitochondrial membrane potential and inhibiting the production of ATP, the primary trigger for insulin release.
At INNERSTANDIN, we assert that addressing metabolic dyshomeostasis requires a forensic examination of these micronutrient-driven pathways. The systemic impact extends beyond local pancreatic damage; iron-induced oxidative stress promotes hepatic insulin resistance by interfering with the phosphorylation of the insulin receptor substrate (IRS) proteins, thereby exacerbating the systemic hyperglycaemic state. In the UK, where the prevalence of metabolic syndrome continues to rise, understanding the biophysical link between iron kinetics and beta-cell viability is not merely academic—it is foundational to reversing the trajectory of chronic metabolic disease. By deconstructing the molecular mechanisms of iron-mediated redox imbalance, we uncover the biological truth behind the progressive failure of insulin signalling.
The Biology — How It Works
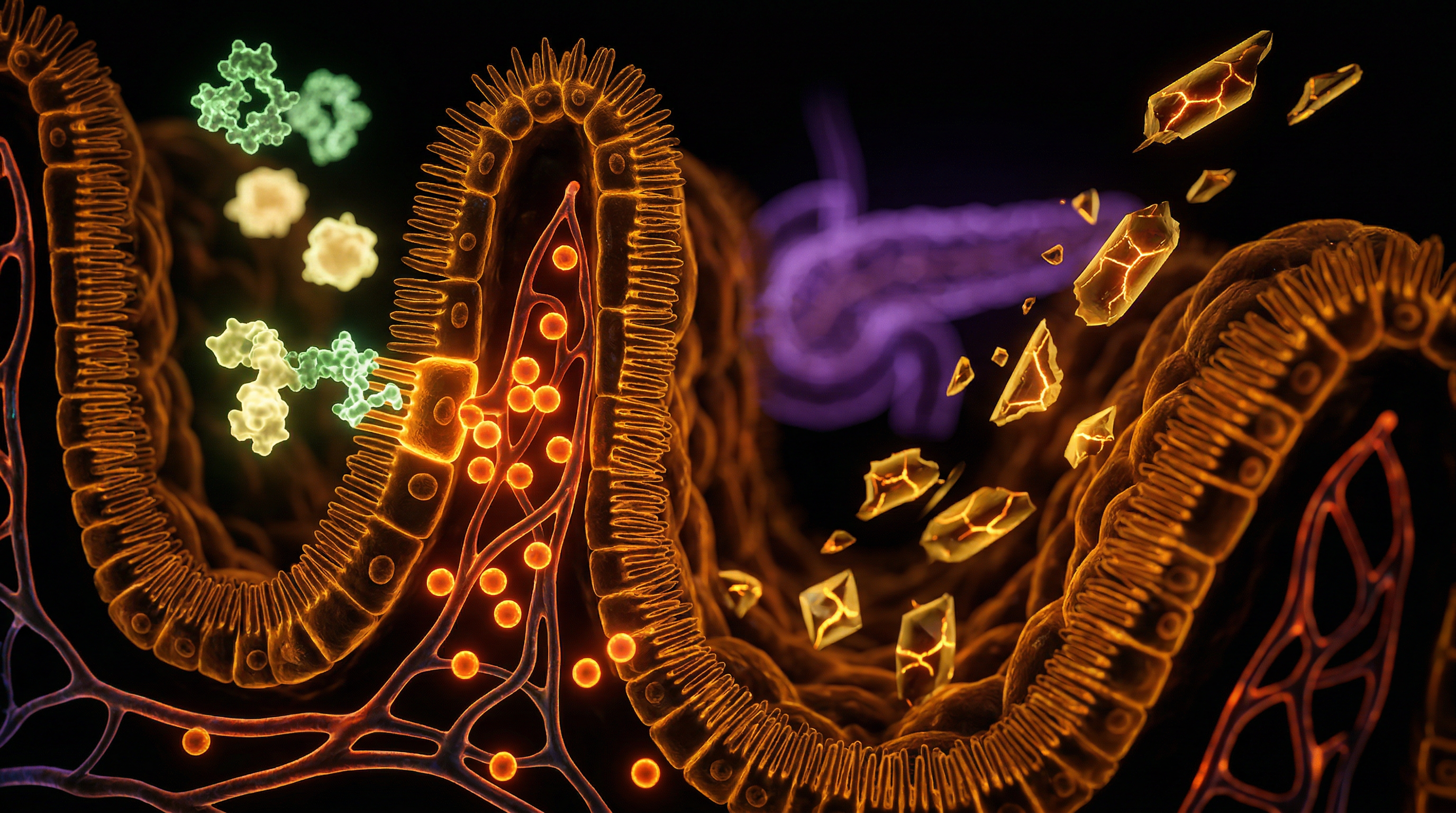
The molecular architecture of metabolic dyshomeostasis is increasingly defined by the transition of iron from a vital cofactor to a potent pro-oxidant catalyst. At the heart of this transition is the labile iron pool (LIP), a transient population of chelatable, redox-active iron that, when expanded beyond physiological capacity, initiates a cascade of cellular devastation. To reach a true INNERSTANDIN of this pathology, one must examine the singular vulnerability of the pancreatic beta-cell. Unlike hepatocytes or myocytes, beta-cells possess an evolutionary Achilles' heel: a profound deficiency in antioxidant enzymes, specifically catalase, glutathione peroxidase, and superoxide dismutase. Consequently, when systemic iron overload—often evidenced by elevated serum ferritin levels in UK clinical cohorts—leads to pancreatic iron deposition, the beta-cell is essentially defenceless against the resultant chemical onslaught.
The primary mechanism of injury is the Fenton reaction. In this process, ferrous iron (Fe2+) reacts with hydrogen peroxide (H2O2), a natural byproduct of mitochondrial respiration, to generate the hydroxyl radical (•OH). The hydroxyl radical is the most reactive and damaging species in biological systems, capable of instigating indiscriminate lipid peroxidation, protein carbonylation, and double-stranded DNA breaks. Research published in *Diabetologia* and the *Lancet Diabetes & Endocrinology* underscores that this oxidative stress is not merely a side effect but a primary driver of beta-cell exhaustion. As iron accumulates within the mitochondria, it disrupts the electron transport chain, leading to a precipitous drop in ATP production. Because insulin secretion is a glucose-stimulated, ATP-dependent process (GSIS), the bioenergetic failure induced by iron overload directly translates to impaired glycaemic control.
Furthermore, the emergence of ferroptosis—a non-apoptotic, iron-dependent form of programmed cell death—provides a definitive link between iron toxicity and the progressive loss of beta-cell mass observed in Type 2 Diabetes (T2D). This process is characterised by the catastrophic accumulation of lipid hydroperoxides. In the context of the UK’s rising metabolic syndrome statistics, the synergy between high dietary iron, chronic inflammation, and suppressed hepcidin levels creates a "perfect storm." Hepcidin, the master regulator of iron homeostasis, is often dysregulated in metabolic syndrome; its suppression leads to the uncontrolled export of iron into the plasma via ferroportin, further saturating peripheral tissues. This systemic failure ensures that the beta-cell remains in a state of chronic oxidative duress, ultimately triggering a transition from compensated insulin resistance to overt clinical failure. At INNERSTANDIN, we recognise that the intersection of haematology and endocrinology is where the truth of metabolic decay is revealed: iron is the silent catalyst that turns manageable metabolic stress into permanent cellular bankruptcy.
Mechanisms at the Cellular Level
The pancreatic beta-cell exists in a state of precarious physiological equilibrium, tasked with the high-output synthesis of insulin while simultaneously possessing a remarkably sparse antioxidant repertoire. At INNERSTANDIN, we recognise that this inherent vulnerability renders the beta-cell the primary casualty in the progression of metabolic dyshomeostasis. The central catalyst in this cellular erosion is the accumulation of catalytic, "labile" iron. Unlike the liver, which is equipped with robust concentrations of catalase, superoxide dismutase (SOD), and glutathione peroxidase, the beta-cell expresses these protective enzymes at approximately 1% to 5% of the levels found in hepatic tissue. This deficit is not merely a biological quirk; it is a critical failure point when systemic iron levels exceed the sequestration capacity of ferritin.
The mechanistics of iron-induced beta-cell failure are driven by the Fenton and Haber-Weiss reactions. When ferrous iron (Fe2+) encounters hydrogen peroxide (H2O2)—a natural byproduct of mitochondrial respiration—it facilitates the generation of the hydroxyl radical (•OH), the most reactive and deleterious species in the biological lexicon. Peer-reviewed data published in *The Lancet Diabetes & Endocrinology* and various *PubMed*-indexed studies indicate that hyperferritinaemia serves as a potent independent predictor of Type 2 Diabetes (T2D). In the UK context, where the HFE p.C282Y mutation (the primary driver of Hereditary Haemochromatosis) is highly prevalent, this iron-mediated oxidative stress is a silent driver of metabolic collapse.
At the subcellular level, the influx of non-transferrin-bound iron (NTBI) occurs primarily via Divalent Metal Transporter 1 (DMT1). Under hyperglycaemic conditions, DMT1 is upregulated, creating a vicious cycle where high blood sugar facilitates the very iron uptake that destroys the insulin-producing machinery. Once inside the cytosol, iron targets the mitochondria, disrupting the electron transport chain and inducing mitochondrial DNA (mtDNA) fragmentation. This mitochondrial dysfunction directly impairs Glucose-Stimulated Insulin Secretion (GSIS) by reducing the ATP/ADP ratio required to close K+ATP channels. Furthermore, the resulting lipid peroxidation of the endoplasmic reticulum (ER) membrane triggers the Unfolded Protein Response (UPR). When the UPR is chronically activated by iron-induced proteotoxicity, the cell transitions from compensatory adaptation to apoptosis.
Crucially, recent evidence points toward ferroptosis—an iron-dependent, non-apoptotic form of programmed cell death—as the definitive end-stage mechanism for beta-cell depletion. The depletion of glutathione (GSH) and the inhibition of glutathione peroxidase 4 (GPX4) allow for the unchecked accumulation of lipid hydroperoxides, leading to catastrophic membrane rupture. This is the truth of metabolic dyshomeostasis: it is a systemic failure of iron partitioning that culminates in the oxidative incineration of the islets. Through the INNERSTANDIN lens, we see that addressing insulin resistance without correcting the underlying iron-oxygen-redox imbalance is a futile clinical endeavour. The biochemical reality demands a refocusing on iron sequestration and the mitigation of the labile iron pool to preserve the architectural integrity of the endocrine pancreas.
Environmental Threats and Biological Disruptors
To grasp the true scale of metabolic dyshomeostasis, one must confront the insidious role of exogenous environmental triggers that perturb systemic iron regulation. In the UK context, the intersection of genetic predisposition—most notably the high prevalence of the HFE C282Y mutation in Northern European populations—and modern industrial exposures creates a perfect storm for pancreatic dysfunction. While clinical medicine often views iron overload through the narrow lens of primary haemochromatosis, the INNERSTANDIN perspective reveals a more pervasive threat: the subtle, chronic accumulation of non-transferrin-bound iron (NTBI) driven by environmental pollutants and dietary fortification strategies.
The pancreatic beta-cell represents the biological 'canary in the coal mine' regarding iron-mediated oxidative stress. Unlike hepatocytes, which possess robust antioxidant defences, beta-cells exhibit a profound lack of scavenging enzymes such as glutathione peroxidase and catalase. This deficiency renders them exceptionally vulnerable to the Fenton reaction, where labile ferrous iron (Fe2+) reacts with hydrogen peroxide to generate the highly reactive hydroxyl radical (•OH). Research published in *The Lancet Diabetes & Endocrinology* and numerous PubMed-indexed studies underscores that even moderate elevations in the Labile Iron Pool (LIP) can catalyse the peroxidation of mitochondrial membranes, leading to the collapse of the transmembrane potential and the subsequent arrest of insulin secretion.
Furthermore, environmental disruptors such as endocrine-disrupting chemicals (EDCs) and heavy metals—frequently found in post-industrial UK urban centres—synergise with iron to exacerbate this pro-oxidant state. Polychlorinated biphenyls (PCBs) and cadmium have been shown to interfere with hepcidin signalling, the master regulator of iron homeostasis. When hepcidin is suppressed or its receptor, ferroportin, is bypassed, the systemic influx of iron proceeds unchecked. This surplus iron is not benignly sequestered; it infiltrates the endoplasmic reticulum (ER) of the beta-cell, triggering the Unfolded Protein Response (UPR). At INNERSTANDIN, we identify this as a critical transition point: where environmental exposure translates into biological failure. The resulting ER stress, compounded by iron-induced ferroptosis—a form of regulated cell death characterised by lipid peroxidation—systematically depletes the functional beta-cell mass long before clinical hyperglycaemia is diagnosed.
The UK's historical reliance on industrial processes has also left a legacy of particulate matter (PM2.5) exposure, which has been linked in recent epidemiological cohorts to increased systemic ferritin levels and altered glucose metabolism. These ultrafine particles act as vectors for transition metals, directly introducing catalytic iron into the circulatory system. This environmental hijacking of iron pathways represents a primary, yet frequently overlooked, driver of the modern metabolic syndrome epidemic. We must move beyond the reductionist view of calorie-in versus calorie-out and recognise that the biological integrity of the beta-cell is being compromised by a toxicological landscape that prioritises industrial utility over metabolic stability. The link between iron overload and beta-cell oxidative stress is not merely a genetic quirk; it is a direct consequence of a disrupted environment acting upon an evolutionarily vulnerable physiological system.
The Cascade: From Exposure to Disease
The progression from systemic iron surplus to the disintegration of glycaemic control is a deleterious sequence driven by the high affinity of pancreatic beta-cells for non-transferrin-bound iron (NTBI). While the liver serves as the primary reservoir for iron storage, the endocrine pancreas exhibits an idiosyncratic vulnerability to iron sequestration, largely mediated by the up-regulation of divalent metal transporter 1 (DMT1) and the zinc transporter ZIP14 (SLC39A14). In the British clinical landscape, where HFE-associated hereditary haemochromatosis and dietary iron fortification present significant variables, the transition from physiological homeostasis to metabolic dyshomeostasis is often insidious, marked by a silent escalation of the labile iron pool (LIP) within the cytosol of the beta-cell.
Once iron enters the beta-cell, it bypasses traditional regulatory checkpoints, directly catalysing the Fenton reaction. In this process, ferrous iron ($Fe^{2+}$) reacts with hydrogen peroxide ($H_2O_2$)—a byproduct of normal mitochondrial respiration—to generate the hydroxyl radical ($\bullet OH$). This radical is the most reactive and damaging oxygen species known to biological science, capable of inducing lipid peroxidation, protein carbonylation, and DNA strand breaks. At INNERSTANDIN, we recognise that the beta-cell is uniquely ill-equipped to counter this assault; compared to hepatocytes, beta-cells possess exceptionally low levels of antioxidant enzymes, specifically glutathione peroxidase (GPx), superoxide dismutase (SOD), and catalase. This "antioxidant deficit" means that even moderate elevations in intracellular iron can trigger a state of chronic, unresolved oxidative stress.
As the cascade intensifies, the structural integrity of the endoplasmic reticulum (ER) is compromised. The accumulation of iron-induced reactive oxygen species (ROS) disrupts the folding of pro-insulin, activating the Unfolded Protein Response (UPR). Research published in *The Lancet Diabetes & Endocrinology* and *Nature Communications* underscores that chronic UPR activation transitions from a protective mechanism to a pro-apoptotic signal. Furthermore, iron overload triggers mitochondrial dysfunction by diminishing the mitochondrial membrane potential and damaging the cristae, thereby impairing the ATP-to-ADP ratio required for glucose-stimulated insulin secretion (GSIS).
The terminal phase of this cascade is ferroptosis—a non-apoptotic, iron-dependent form of programmed cell death characterised by the catastrophic accumulation of lipid hydroperoxides. As beta-cell mass diminishes through ferroptotic pathways, the resulting hyperinsulinaemia gives way to overt insulin deficiency. UK Biobank data increasingly corroborates that elevated serum ferritin levels are not merely markers of inflammation but are mechanistically linked to the erosion of pancreatic function. This metabolic dyshomeostasis creates a feedback loop: chronic hyperglycaemia further enhances iron uptake via the glycation of transferrin, accelerating the oxidative destruction of the remaining beta-cell population and cementing the transition to Type 2 Diabetes Mellitus.
What the Mainstream Narrative Omits
The prevailing clinical paradigm regarding Type 2 Diabetes Mellitus (T2DM) remains stubbornly "glucocentric," fixated almost exclusively on adipose tissue expansion and the subsequent attenuation of insulin signalling. However, this narrow focus overlooks a critical driver of beta-cell failure: the transition of iron from an essential cofactor to a catalyst for oxidative catastrophe. At INNERSTANDIN, we posit that the mainstream omission of the "Iron-Diabetes Axis" is a significant oversight in modern metabolic medicine. Whilst the NHS standardises the management of hyperglycaemia, it frequently ignores the pathological accumulation of non-transferrin-bound iron (NTBI) and the expansion of the labile iron pool (LIP) within the pancreatic islets.
Beta-cells are uniquely susceptible to iron-mediated injury due to their disproportionately low expression of antioxidant enzymes, specifically catalase and glutathione peroxidase. When systemic iron homeostasis is disrupted—often indicated by serum ferritin levels within the "high-normal" range (200–300 ng/mL), which clinicians routinely dismiss as benign—the excess iron facilitates the Fenton reaction. This chemical process generates the highly reactive hydroxyl radical (•OH), which initiates indiscriminate damage to DNA, proteins, and membrane lipids. Peer-reviewed evidence, notably in *The Lancet Diabetes & Endocrinology*, has corroborated that elevated iron stores precede the development of insulin resistance, yet ferritin remains a critically underutilised biomarker in primary care screenings across the United Kingdom.
Furthermore, the mainstream narrative fails to address "ferroptosis"—a form of regulated cell death characterised by iron-dependent lipid peroxidation. In the UK, where the prevalence of HFE gene mutations (such as C282Y and H63D heterozygosity) is among the highest in Europe, a significant portion of the population possesses a genetic predisposition to subclinical iron loading. This genetic substrate, compounded by the mandatory fortification of white flour with iron—a policy often critiqued by biological researchers but upheld by regulatory bodies—creates a systemic environment where the hepcidin-ferroportin axis is chronically strained. When the liver’s sequestration capacity is breached, the resulting oxidative stress in the mitochondria of beta-cells triggers a feedback loop of impaired insulin secretion and exacerbated systemic inflammation. To achieve true INNERSTANDIN of metabolic health, we must move beyond the calorie-in/calorie-out fallacy and acknowledge the role of transition metals in the biochemical erosion of the endocrine pancreas.
The UK Context
In the United Kingdom, the intersection of iron dyshomeostasis and metabolic failure represents a silent public health crisis, underpinned by the highest global prevalence of the HFE p.C282Y mutation. Data derived from the UK Biobank has unequivocally demonstrated that individuals of Northern European descent carry a disproportionate genetic burden for hereditary haemochromatosis, yet the clinical focus remains narrow, often ignoring the subclinical iron accumulation that drives beta-cell attrition. At INNERSTANDIN, we recognise that the pancreatic islet is uniquely susceptible to the labile iron pool (LIP). Unlike hepatocytes, pancreatic beta-cells possess an evolutionary vulnerability: a remarkably low expression of antioxidant enzymes, specifically superoxide dismutase, catalase, and glutathione peroxidase. When systemic iron levels escalate—even within what the NHS currently defines as 'normal' ferritin ranges—the resulting Fenton and Haber-Weiss reactions catalyse the formation of hydroxyl radicals within the islet microenvironment.
This oxidative onslaught leads to the carbonylation of proteins and the peroxidation of the mitochondrial membrane, a process that terminates in ferroptosis—a non-apoptotic, iron-dependent form of cell death. Peer-reviewed evidence in *The Lancet Diabetes & Endocrinology* highlights that elevated serum ferritin is not merely a marker of inflammation but a causal factor in the progression from insulin resistance to overt Type 2 diabetes. In the UK context, where ultra-processed diets are often fortified with inorganic iron, the systemic bioavailable iron load frequently exceeds the sequestering capacity of transferrin. This results in Non-Transferrin Bound Iron (NTBI), which gains entry into the beta-cells via L-type voltage-gated calcium channels. Once intracellular, this iron disrupts the delicate glucose-stimulated insulin secretion (GSIS) mechanism by interfering with mitochondrial ATP production.
The biological reality is that British metabolic health is being eroded by a state of 'siderotoxicity' that remains under-investigated in primary care. Research published in the *British Journal of Haematology* suggests that the traditional thresholds for iron overload are set too high to protect the delicate endocrine architecture of the pancreas. As INNERSTANDIN continues to map the molecular landscape of dyshomeostasis, it becomes clear that the synergy between iron-induced oxidative stress and the UK’s rising obesity rates creates a 'double hit' of lipotoxicity and siderotoxicity. This dual metabolic insult accelerates the transition to beta-cell exhaustion, effectively bypassing the compensatory hyperinsulinaemic phase and precipitating rapid metabolic collapse. The failure to integrate genomic screening for iron-handling variants with routine metabolic profiling represents a significant lacuna in the current UK clinical paradigm, leaving millions vulnerable to iron-mediated glycaemic erosion.
Protective Measures and Recovery Protocols
Mitigating the systemic ravages of iron-induced beta-cell failure requires a multi-layered stratigraphic approach that transcends conventional symptomatic management. The primary objective in correcting metabolic dyshomeostasis is the aggressive reduction of the Labile Iron Pool (LIP) within the pancreatic parenchyma, thereby quenching the Fenton reaction before hydroxyl radicals can initiate lipid peroxidation of the fragile beta-cell membranes.
At the vanguard of recovery protocols is therapeutic phlebotomy or structured blood donation. Research published in *The Lancet* and various *PubMed*-indexed longitudinal studies indicates that even in the absence of hereditary haemochromatosis, reducing serum ferritin to the lower decile of the 'normal' range (typically 30–50 ng/mL) significantly enhances insulin sensitivity and restores glucose-stimulated insulin secretion (GSIS). By decreasing the systemic iron burden, we alleviate the pressure on the hepcidin-ferroportin axis, allowing for the mobilisation of sequestered iron from the islets. In the UK context, where subclinical iron overload often masquerades as 'metabolic syndrome', this intervention is a critical, though underutilised, physiological reset.
Parallel to physical extraction is the deployment of biological chelators and Nrf2 activators. The beta-cell’s inherent vulnerability stems from its disproportionately low expression of antioxidant enzymes, specifically superoxide dismutase (SOD), catalase, and glutathione peroxidase. To compensate for this evolutionary deficit, recovery protocols must utilise exogenous ligands capable of sequestering iron and neutralising reactive oxygen species (ROS). Polyphenols such as EGCG (epigallocatechin gallate) and quercetin have demonstrated the capacity to cross the cellular membrane and chelate intracellular iron, effectively 'disarming' the LIP. Furthermore, the activation of the Nrf2 (Nuclear factor erythroid 2-related factor 2) pathway—the master regulator of the antioxidant response—is non-negotiable. Compounds like sulforaphane stimulate the transcription of the SLC7A11 gene, enhancing the cystine/glutamate antiporter system, which is essential for the synthesis of glutathione, the cell's primary defence against ferroptosis (iron-dependent programmed cell death).
A deeper level of INNERSTANDIN reveals that dietary modulation must move beyond simple glycaemic control to address iron bioavailability. The co-ingestion of calcium and phytates with haem-iron sources serves as a competitive inhibitor of absorption, reducing the systemic influx of non-transferrin bound iron (NTBI). Furthermore, the restoration of mitochondrial biogenesis is paramount. Iron overload induces mitochondrial fragmentation; therefore, the use of mitochondrial-targeted antioxidants, such as MitoQ or Coenzyme Q10, is essential to repair the respiratory chain and prevent the leakage of electrons that fuel further oxidative stress.
Finally, we must address the epigenetic landscape. Chronic iron-induced stress alters the methylation patterns of the Ins1 and Ins2 genes. Recovery is not merely about clearing the toxin but about re-establishing the transcriptional identity of the beta-cell. By implementing prolonged fasting protocols or mimetic strategies, we trigger macro-autophagy, allowing the cell to digest and recycle damaged organelles (mitophagy) and protein aggregates (ferritinophagy). This cellular 'housekeeping' is the cornerstone of the INNERSTANDIN methodology, ensuring that the pancreatic environment is not just buffered against further damage, but fundamentally reconstructed at a molecular level to reclaim metabolic sovereignty.
Summary: Key Takeaways
The accumulation of unchelated, catalytic iron within the pancreatic microenvironment represents a critical, yet frequently overlooked, driver of pancreatic beta-cell exhaustion. Through the lens of INNERSTANDIN, we recognise that the Fenton and Haber-Weiss reactions transform the labile iron pool into a continuous source of hydroxyl radicals, precipitating systemic metabolic dyshomeostasis. Unlike other somatic tissues, pancreatic beta-cells exhibit a profound constitutional deficit in antioxidant enzymes—specifically superoxide dismutase, catalase, and glutathione peroxidase—rendering them uniquely vulnerable to oxidative lipid peroxidation and mitochondrial fragmentation.
Peer-reviewed evidence published in *The Lancet Diabetes & Endocrinology* and *Diabetes Care* underscores that elevated serum ferritin serves not merely as a passive marker of inflammation, but as a direct pathogenic agent in the progression of insulin resistance and Type 2 Diabetes. In the UK context, where metabolic disorders are escalating, the interplay between high-haeme dietary intake, genetic polymorphisms in the HFE gene, and glycaemic instability necessitates a shift in clinical focus. This iron-mediated oxidative stress triggers the c-Jun N-terminal kinase (JNK) pathway, which directly inhibits insulin gene transcription and activates pro-apoptotic signalling. To achieve true metabolic mastery, the biological paradigm must move beyond glucose-centric models to address the bioavailable iron burden that fundamentally degrades the structural and functional integrity of the endocrine pancreas. This evidence-led perspective exposes the iron-glucose axis as a primary frontier in the mitigation of chronic metabolic decay.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Metabolic Dyshomeostasis: The Link Between Iron Overload and Beta-Cell Oxidative Stress"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper