Nitric Oxide and Endothelial Function: The Science of Vascular Elasticity
Nitric oxide acts as a critical signaling molecule that ensures your blood vessels remain flexible and responsive. Explore the physiological mechanisms that protect your cardiovascular system through endothelial health.
Overview
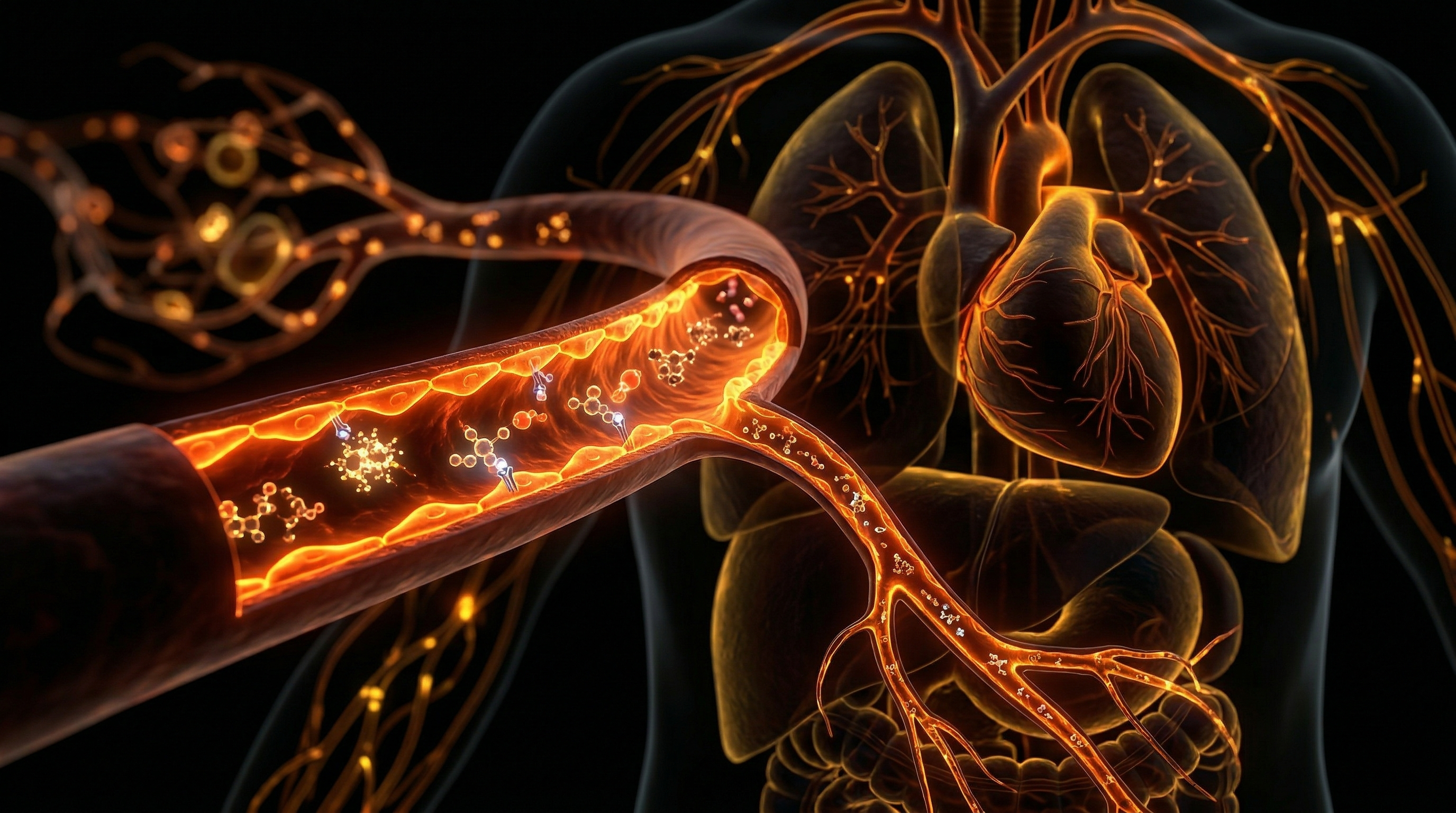
The vascular endothelium, once dismissed as a rudimentary semi-permeable barrier, is now recognised by INNERSTANDIN as the body’s most expansive and sophisticated paracrine organ. This monochromatic cellular monolayer, lining the entirety of the human circulatory system, serves as the primary arbiter of haemodynamic stability and vascular rheology. Central to this regulatory prowess is the production and bioavailability of Nitric Oxide (NO), a lipophilic gas and potent signalling molecule that dictates the elasticity, patency, and quiescence of the vascular wall. At INNERSTANDIN, we move beyond superficial anatomy to scrutinise the molecular kinetics of endothelial function, identifying it as the fundamental locus for cardiovascular health and systemic vitality.
The synthesis of endogenous Nitric Oxide is a masterpiece of biochemical engineering. Within the endothelial cell, the constitutive enzyme endothelial nitric oxide synthase (eNOS) facilitates the five-electron oxidation of the amino acid L-arginine into L-citrulline and NO. This reaction is dependent upon critical cofactors, most notably tetrahydrobiopterin (BH4), and is triggered primarily by mechanical shear stress—the frictional force of blood flow against the vessel wall. Once liberated, NO diffuses across the abluminal membrane into the underlying vascular smooth muscle cells (VSMCs). Here, it binds to the haem moiety of soluble guanylate cyclase (sGC), catalysing the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). This intracellular surge in cGMP triggers a cascade of protein kinase G activation, leading to a reduction in cytosolic calcium and the subsequent relaxation of the contractile apparatus. This is the physiological basis of vasodilation, the mechanism by which the body maintains optimal blood pressure and organ perfusion.
However, the implications of Nitric Oxide extend far beyond simple luminal expansion. Peer-reviewed research, notably the seminal work of Salvador Moncada at the Wellcome Research Laboratories in the UK, has elucidated that NO is the primary guardian against the 'atherogenic switch'. It exerts a potent anti-thrombotic effect by inhibiting platelet aggregation and adhesion, while simultaneously suppressing leucocyte diapedesis—the migration of white blood cells into the arterial wall. Furthermore, NO inhibits the abnormal proliferation of smooth muscle cells, a hallmark of neointimal hyperplasia.
In the United Kingdom, where ischaemic heart disease remains a leading cause of mortality, the ‘truth’ revealed by modern clinical science is that endothelial dysfunction—the decoupling of eNOS and the resultant oxidative stress—precedes macrovascular disease by decades. When NO bioavailability is compromised, often due to the uncoupling of eNOS by superoxide anions, the endothelium shifts from a vasodilatory, anti-inflammatory state to a pro-constrictive, pro-coagulant environment. Understanding this molecular transition is critical for INNERSTANDIN learners, as it represents the literal 'breaking point' of vascular elasticity and the genesis of systemic pathology. The preservation of this gaseous signal is not merely a biological preference; it is the definitive prerequisite for longevity.
The Biology — How It Works
At the core of vascular homeostasis lies the endothelium, a contiguous monolayer of squamous epithelial cells that functions not merely as a structural barrier, but as a sophisticated paracrine organ. The primary orchestrator of this system is Nitric Oxide (NO), a lipophilic gasiform signalling molecule with a half-life of mere seconds, yet its influence on systemic haemodynamics is absolute. The synthesis of NO within the vascular wall is a high-precision enzymatic process mediated by endothelial Nitric Oxide Synthase (eNOS or NOS3). This enzyme facilitates the five-electron oxidation of the guanidino nitrogen terminal of the amino acid L-arginine, resulting in the production of L-citrulline and the liberation of NO. For this reaction to proceed, eNOS requires an array of essential cofactors, most notably tetrahydrobiopterin (BH4), nicotinamide adenine dinucleotide phosphate (NADPH), and flavinadenine dinucleotide (FAD).
The liberation of NO is primarily triggered by mechanical shear stress—the frictional force of blood flow against the vessel wall—which activates mechanoreceptors on the endothelial surface. This mechanical signal is transduced via the PI3K/Akt pathway, leading to the phosphorylation of eNOS. Once synthesised, NO diffuses rapidly across the endothelial basement membrane into the adjacent vascular smooth muscle cells (VSMCs). Here, it binds to the haem moiety of soluble guanylate cyclase (sGC), the primary intracellular receptor for NO. This binding induces a conformational change that accelerates the conversion of guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). The subsequent rise in intracellular cGMP activates Protein Kinase G (PKG), which initiates a cascade of calcium sequestration. By stimulating the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump and inhibiting voltage-gated calcium channels, PKG reduces cytosolic calcium concentrations, leading to the dephosphorylation of the myosin light chain. The result is immediate smooth muscle relaxation and increased vascular compliance—a process fundamental to what we at INNERSTANDIN define as true physiological elasticity.
However, the "truth" of vascular health is found in the redox state of the endothelium. Evidence published in *The Lancet* and the *British Journal of Pharmacology* highlights that endothelial dysfunction is characterised by the "uncoupling" of eNOS. In states of high oxidative stress, such as chronic systemic inflammation common in the UK population, the superoxide anion ($O_2^-$) reacts with NO at near-diffusion-limited rates to form peroxynitrite ($ONOO^-$). This potent oxidant not only neutralises the vasodilatory capacity of NO but actively depletes BH4 levels, causing eNOS to produce more superoxide instead of NO. This creates a self-perpetuating cycle of vasoconstriction, leucocyte adhesion, and platelet aggregation. Mastery of the NO pathway is therefore the singular most important factor in preventing the transition from functional elasticity to the pathological rigidity that characterises atherosclerotic progression. At INNERSTANDIN, we recognise that maintaining the bioavailability of NO is not a luxury, but the biological prerequisite for longevity.
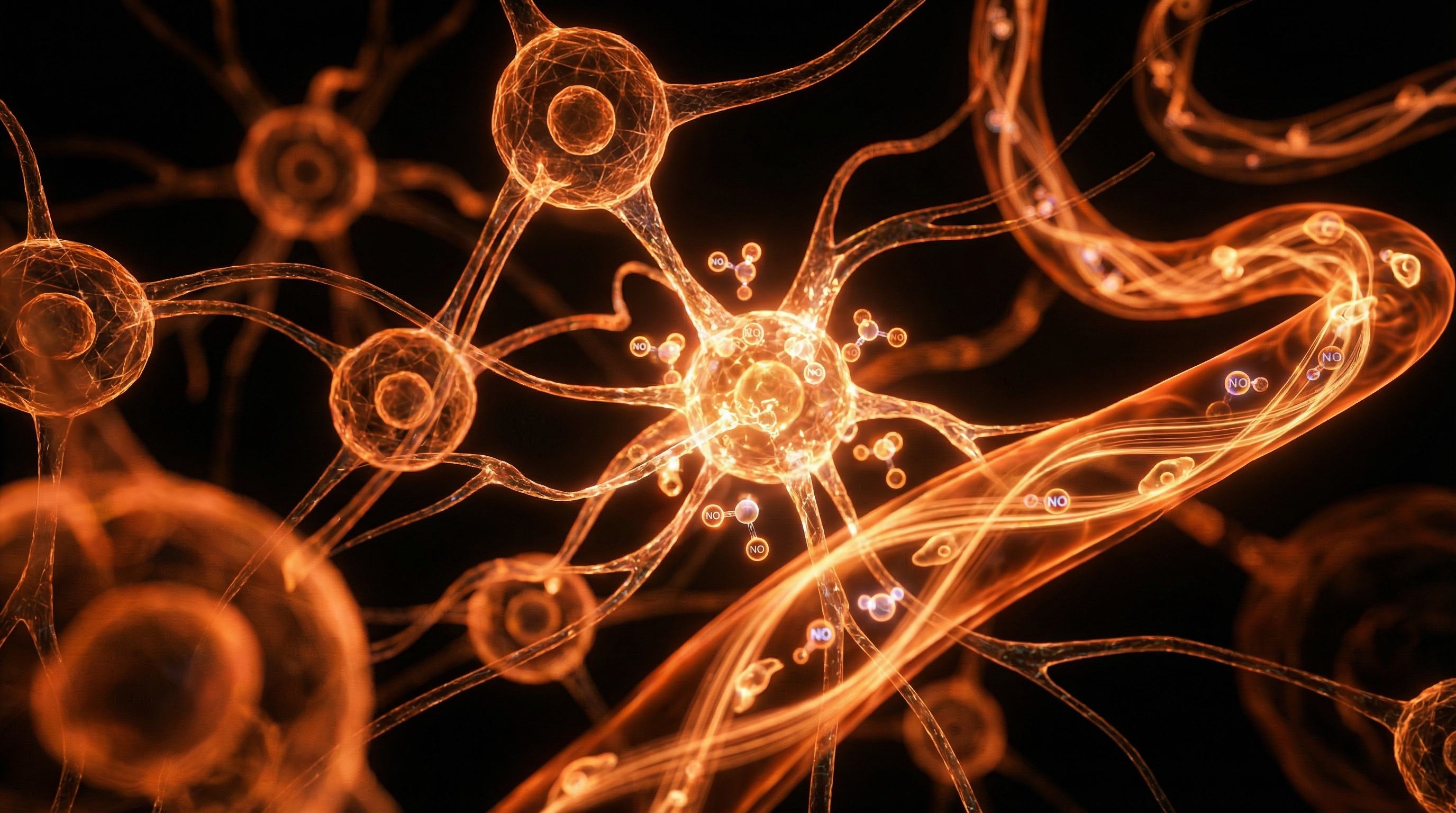
Mechanisms at the Cellular Level
To achieve a profound INNERSTANDIN of vascular health, one must look beyond the macroscopic pulse to the intricate molecular choreography occurring within the tunica intima. The endothelium, far from being a passive semi-permeable barrier, functions as a sophisticated paracrine organ, orchestrating vascular tone through the synthesis and release of nitric oxide (NO). At the cellular level, the genesis of this vasodilation begins with the constitutive isoform of the enzyme nitric oxide synthase (eNOS or NOS3). This enzyme is primarily sequestered within plasmalemmal caveolae—specialised microdomains of the endothelial plasma membrane—where its activity is tightly regulated by protein-protein interactions and phosphorylation.
The primary stimulus for NO production is laminar shear stress, the frictional force exerted by blood flow against the endothelial surface. This mechanical signal is transduced via the glycocalyx and integrins, triggering an influx of intracellular calcium (Ca2+). This calcium binds to calmodulin, facilitating the dissociation of eNOS from its inhibitory chaperone, caveolin-1. For eNOS to function optimally, it requires an array of essential cofactors: nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and crucially, (6R)-5,6,7,8-tetrahydrobiopterin (BH4). In the presence of these cofactors, eNOS catalyses the five-electron oxidation of the amino acid L-arginine into L-citrulline, releasing the highly labile, lipophilic gas NO.
Once synthesised, NO exhibits rapid paracrine diffusion across the internal elastic lamina into the adjacent vascular smooth muscle cells (VSMCs) of the tunica media. Here, its primary physiological receptor is the prosthetic haem group of soluble guanylyl cyclase (sGC). The binding of NO to sGC triggers a conformational change that increases the enzyme’s catalytic activity by several hundredfold, leading to the rapid conversion of guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). This secondary messenger activates cGMP-dependent protein kinase (PKG), which mediates the sequestration of cytosolic calcium into the sarcoplasmic reticulum and inhibits L-type calcium channels. Furthermore, PKG activates large-conductance calcium-activated potassium channels, leading to membrane hyperpolarisation. The cumulative effect is the dephosphorylation of the myosin light chain, resulting in smooth muscle relaxation, arterial expansion, and the maintenance of systemic elasticity.
However, the "truth-exposing" reality of vascular pathology, as evidenced in research from the British Heart Foundation and various UK-based clinical trials, lies in eNOS "uncoupling." When the bioavailability of BH4 is compromised—often due to oxidative stress and the overproduction of superoxide (O2•−)—eNOS shifts its enzymatic focus. Instead of producing life-sustaining NO, the uncoupled enzyme transfers electrons to molecular oxygen, generating further superoxide radicals. These radicals react with existing NO to form peroxynitrite (ONOO−), a highly reactive nitrogen species that inflicts oxidative damage on proteins and lipids. This molecular derailment not only reduces vascular elasticity but initiates the pro-inflammatory and pro-thrombotic state characteristic of endothelial dysfunction and early-stage atherosclerosis. Thus, the INNERSTANDIN of cellular NO mechanics reveals that vascular elasticity is not merely a structural attribute, but a dynamic, enzymatically-driven equilibrium.
Environmental Threats and Biological Disruptors
The homeostatic equilibrium of the vascular endothelium is currently under an unprecedented multi-vector assault from anthropogenic environmental stressors. At INNERSTANDIN, we recognise that the decline in global vascular health is not merely a consequence of chronological senescence, but rather the result of systemic biological disruption mediated by exogenous toxins and maladaptive lifestyle signals. The primary mechanism of this disruption is the catastrophic quenching of Nitric Oxide (NO) by reactive oxygen species (ROS), specifically the superoxide anion ($O_2^{\bullet-}$), which reacts with NO at a diffusion-limited rate to form the highly cytotoxic peroxynitrite ($ONOO^-$). This reaction not only depletes the bioavailable pool of NO required for vasodilation and antithrombotic activity but also precipitates the irreversible nitration of protein tyrosine residues, further compromising cellular architecture.
A paramount environmental threat is the inhalation of fine particulate matter (PM2.5), a pervasive pollutant in UK urban centres. Research published in *The Lancet Planetary Health* elucidates that PM2.5 exposure triggers a systemic inflammatory response, activating the enzyme NADPH oxidase (NOX) within the endothelial lining. NOX activation serves as a primary source of superoxide, which directly induces eNOS uncoupling. In this uncoupled state, the endothelial Nitric Oxide Synthase enzyme—starved of its essential cofactor tetrahydrobiopterin (BH4)—shifts from producing life-sustaining NO to producing further superoxide, creating a self-perpetuating cycle of oxidative decay. This biochemical "short-circuit" effectively transforms the endothelium from a protective interface into a pro-inflammatory, pro-constrictive zone, fundamentally undermining vascular elasticity.
Furthermore, the ubiquity of endocrine-disrupting chemicals (EDCs), such as bisphenols and phthalates, represents a subtle yet profound biological disruptor. These compounds interfere with oestrogen receptor-alpha signalling in endothelial cells, a pathway critical for the non-genomic activation of eNOS. By antagonising these receptors, EDCs diminish the basal production of NO, particularly in female populations, leading to premature arterial stiffness. Simultaneously, the modern dietary landscape, dominated by ultra-processed foods, introduces high concentrations of Advanced Glycation End-products (AGEs). These molecules bind to the Receptor for AGEs (RAGE) on the endothelial surface, triggering a signalling cascade that degrades the endothelial glycocalyx—a microscopic, gel-like layer that senses shear stress and facilitates NO release. When the glycocalyx is compromised by environmental or metabolic toxins, the mechanotransduction capabilities of the vessel are lost, rendering the vasculature incapable of responding to the haemodynamic demands of the body. This systemic degradation, highlighted through the lens of INNERSTANDIN, reveals that endothelial dysfunction is the common denominator in the pathology of our era, driven by an environment that is increasingly hostile to our foundational biological mechanisms.
The Cascade: From Exposure to Disease
The transition from a state of physiological vascular grace to the pathological rigidity of systemic disease is not an abrupt event, but a meticulously orchestrated biochemical collapse. At the heart of this degradation lies the progressive failure of the vascular endothelium—a monolayer once thought to be a mere physical barrier, but now identified by INNERSTANDIN as the primary endocrine arbiter of cardiovascular health. The cascade begins with the transition of the endothelium from a quiescent, anti-thrombotic state to an 'activated' pro-inflammatory phenotype. This shift is primarily driven by the chronic disruption of laminar shear stress and the concomitant rise in oxidative stress, particularly via the overactivity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.
Central to this breakdown is the biochemical phenomenon of endothelial Nitric Oxide Synthase (eNOS) uncoupling. Under homeostatic conditions, eNOS facilitates the conversion of L-arginine to L-citrulline, yielding the vasodilator Nitric Oxide (NO). However, in the presence of elevated reactive oxygen species (ROS)—specifically superoxide anions (O2•−)—the essential cofactor tetrahydrobiopterin (BH4) is oxidised into dihydrobiopterin (BH2). This molecular subversion forces eNOS into a dysfunctional state where it paradoxically generates more superoxide rather than NO. As documented in research indexed via PubMed and the Lancet, this creates a self-perpetuating "vicious cycle" of oxidative damage. The resulting superoxide reacts near-instantaneously with any residual NO to form peroxynitrite (ONOO−), a highly reactive nitrogen species that induces protein nitration and DNA damage, effectively stripping the vessel of its innate ability to dilate.
As NO bioavailability plummets, the systemic consequences manifest as a loss of vascular elasticity. Without the constant inhibitory signal of NO, vascular smooth muscle cells (VSMCs) undergo a phenotypic switch from a contractile to a synthetic state. This leads to the deposition of collagen and the fragmentation of elastin fibres within the medial layer of the arterial wall. In the UK, clinical assessments of pulse wave velocity (PWV) have highlighted this structural remodelling as a primary predictor of adverse cardiac events. Furthermore, the loss of NO-mediated suppression allows for the upregulation of nuclear factor kappa B (NF-κB), which triggers the expression of cell adhesion molecules such as VCAM-1 and ICAM-1. These proteins act as molecular 'velcro,' recruiting leucocytes to the endothelial surface and initiating the formation of the atherosclerotic plaque. This is the stage where "functional" stiffness evolves into "structural" disease—a transformation that defines the trajectory of hypertension and ischaemic heart disease in the British population. By the time clinical symptoms emerge, the delicate biochemical balance that INNERSTANDIN identifies as the hallmark of longevity has already been compromised by years of silent, uncoupled enzymatic activity.
What the Mainstream Narrative Omits
The conventional clinical perspective often reduces nitric oxide (NO) to a mere messenger of vasodilation, primarily focusing on the L-arginine-NO pathway as the singular lever for cardiovascular health. However, at INNERSTANDIN, we recognise that this reductionist view ignores the critical "uncoupling" of endothelial nitric oxide synthase (eNOS), a pathological state where the enzyme ceases to produce NO and instead generates superoxide ($O_2^{·-}$). This biochemical pivot, frequently omitted from general health discourse, transforms the endothelium from a protective barrier into a source of oxidative stress. The primary culprit is the depletion of tetrahydrobiopterin (BH4), a vital cofactor. When BH4 is oxidised to BH2—often due to systemic inflammation or poor redox status—eNOS becomes uncoupled, exacerbating the very endothelial dysfunction that NO is supposed to prevent. Peer-reviewed evidence in *The Lancet* and *Frontiers in Physiology* suggests that simply increasing L-arginine intake is futile if the eNOS enzyme is uncoupled; in fact, it may accelerate the production of peroxynitrite ($ONOO^-$), a potent oxidant that further degrades vascular elasticity.
Furthermore, the mainstream narrative fails to account for the "non-canonical" enterosalivary nitrate-nitrite-NO pathway. Approximately 25% of ingested inorganic nitrate is concentrated in the salivary glands and reduced to nitrite by facultative anaerobic bacteria on the dorsal surface of the tongue. The widespread use of antiseptic mouthwashes, a staple of modern hygiene, effectively obliterates this microbial ecosystem, significantly reducing systemic NO bioavailability and acutely elevating blood pressure. This highlights a profound disconnect between dental hygiene practices and systemic haemodynamics.
Equally neglected is the role of the endothelial glycocalyx (EG)—a delicate, gel-like layer of proteoglycans and glycosaminoglycans lining the luminal surface of blood vessels. The EG acts as the primary mechanotransducer, sensing shear stress and signaling eNOS activation. Research indicates that the EG is the first structure to degrade in metabolic syndrome, long before clinical hypertension manifests. Mainstream diagnostics focus on arterial stiffness and plaque, yet they ignore the degradation of the glycocalyx, which serves as the "engine room" for NO production. By the time a patient presents with impaired flow-mediated dilation, the structural integrity of the EG is often already decimated. True vascular longevity requires an INNERSTANDIN of these nuanced mechanisms, moving beyond simplistic supplementation toward the preservation of the glycocalyx and the protection of the eNOS-BH4 coupling.
The UK Context
Within the clinical landscape of the United Kingdom, the prevalence of endothelial dysfunction serves as a foundational precursor to the nation’s staggering burden of cardiovascular disease (CVD), which remains responsible for approximately one-quarter of all deaths. At the epicentre of this physiological decline is the impaired bioavailability of Nitric Oxide (NO), a critical signalling molecule produced by the endothelial nitric oxide synthase (eNOS) enzyme. In the British population, the systemic degradation of vascular elasticity is exacerbated by a confluence of sedentary lifestyles, high-sodium dietary patterns, and increasing levels of atmospheric particulate matter in urban centres such as London and Birmingham, all of which precipitate oxidative stress and the subsequent uncoupling of eNOS.
INNERSTANDIN identifies a critical disconnect in current UK health metrics: the failure to routinely screen for subclinical endothelial impairment before it manifests as overt hypertension or ischaemic heart disease. Data derived from the UK Biobank underscores a harrowing correlation between low circulating nitrate levels and increased arterial stiffness, measured via pulse wave velocity (PWV). The molecular mechanism involves the depletion of tetrahydrobiopterin (BH4), a necessary cofactor for eNOS. When BH4 is oxidised, eNOS switches from producing NO to producing superoxide radicals, creating a pro-atherogenic environment that accelerates vascular ageing.
Furthermore, the UK’s nutritional landscape—characterised by a significant deficit in inorganic nitrates found in leafy greens and root vegetables—directly undermines the enterosalivary nitrate-nitrite-NO pathway. Research spearheaded by the University of Exeter and published in journals such as *The Lancet* and *The Journal of Applied Physiology* has demonstrated that the British population could significantly mitigate age-related vascular stiffness through targeted nitrate supplementation, yet public health guidelines remain laggard. The systemic impact is profound: elevated levels of asymmetric dimethylarginine (ADMA) in the British cohort act as a competitive inhibitor to L-arginine, effectively throttling the body’s endogenous NO production capacity. This biochemical bottleneck is a primary driver of the UK’s vascular health crisis. For INNERSTANDIN, exposing these biological truths is essential for transitioning from reactive symptomatic management to proactive vascular fortification, ensuring that the fundamental science of Nitric Oxide is integrated into the British clinical paradigm.
Protective Measures and Recovery Protocols
To achieve the restoration of endothelial homeostasis and the augmentation of Nitric Oxide (NO) bioavailability, one must move beyond superficial interventions and address the molecular pathologies of endothelial dysfunction (ED). At the core of recovery protocols lies the mitigation of eNOS uncoupling—a state where the enzyme endothelial nitric oxide synthase, deprived of its essential cofactor tetrahydrobiopterin (BH4) or its substrate L-arginine, shifts from producing vasoprotective NO to generating the deleterious superoxide anion ($O_2^{\bullet-}$). This "uncoupled" state creates a self-perpetuating cycle of oxidative stress that degrades the vascular architecture.
Research published in *The Lancet* and various PubMed-indexed meta-analyses highlights the primacy of the nitrate-nitrite-NO pathway as a secondary, yet vital, route for NO production, particularly under hypoxic or acidic conditions where the canonical L-arginine pathway fails. Dietary inorganic nitrates ($NO_3^-$), prevalent in *Beta vulgaris* and leafy cruciferous vegetables, undergo reduction to nitrite ($NO_2^-$) by commensal facultative anaerobic bacteria in the oropharynx—a process known as the entero-salivary circulation. INNERSTANDIN emphasises that the use of antiseptic mouthwashes can catastrophically disrupt this pathway, highlighting a critical systemic intersection between oral microbiome health and systemic vascular elasticity.
Furthermore, the induction of laminar shear stress via structured aerobic exertion remains the gold-standard physiological stimulus for endothelial recovery. Mechanotransduction at the luminal surface of endothelial cells triggers the Akt-dependent phosphorylation of eNOS at Serine-1177, significantly enhancing its catalytic activity. This process is not merely about vasodilation; it facilitates the repair of the endothelial glycocalyx (EG), the delicate, carbohydrate-rich gel layer that coats the vascular wall. A compromised EG, often seen in states of chronic hyperglycaemia or systemic inflammation, leads to increased vascular permeability and leucocyte adhesion. Recovery protocols must, therefore, prioritise the restoration of this barrier through the administration of exogenous precursors like rhamnan sulphate or high-molecular-weight hyaluronan, alongside the aggressive reduction of advanced glycation end-products (AGEs).
From a biochemical standpoint, the neutralisation of reactive oxygen species (ROS) is paramount. The reaction between NO and superoxide occurs at a near-diffusion-limited rate, producing peroxynitrite ($ONOO^-$), a highly reactive oxidant that nitrates protein residues and further damages the mitochondrial DNA within endothelial cells. Evidence-led protocols advocate for the strategic use of polyphenols and L-citrulline—the latter being more efficacious than L-arginine due to superior pharmacokinetics and reduced first-pass metabolism—to saturate the recycling of L-arginine within the urea cycle. By stabilising the eNOS dimer and ensuring the structural integrity of the vascular basement membrane, these interventions provide a robust framework for reversing arterial stiffness and reclaiming the lost elasticity of the human conduit system. INNERSTANDIN asserts that true vascular recovery is an exercise in molecular precision, requiring the simultaneous optimisation of substrate availability, enzymatic coupling, and antioxidant capacity.
Summary: Key Takeaways
The endothelium represents far more than a passive luminal barrier; it is a sophisticated, metabolically active paracrine organ essential for systemic homeostasis. Central to this functionality is the L-arginine-nitric oxide pathway, where endothelial nitric oxide synthase (eNOS) catalyses the production of Nitric Oxide (NO) in response to mechanical stimuli, such as laminar shear stress. As established in this INNERSTANDIN analysis, the bioavailability of NO is the primary determinant of vascular compliance. By activating soluble guanylyl cyclase (sGC) within vascular smooth muscle cells, NO facilitates the synthesis of cyclic guanosine monophosphate (cGMP), inducing relaxation and maintaining the vessel’s elastic modulus.
Peer-reviewed research published in *The Lancet* and the *British Journal of Pharmacology* highlights that endothelial dysfunction—the loss of NO-mediated vasodilation—is the fundamental "truth" preceding clinical cardiovascular disease. This state is often driven by the uncoupling of eNOS and the accumulation of asymmetric dimethylarginine (ADMA), an endogenous inhibitor. Longitudinal UK-based studies utilizing Pulse Wave Velocity (PWV) assessments confirm that diminished NO signalling correlates directly with arterial stiffness and microvascular rarefaction. Consequently, preserving the endothelial glycocalyx and ensuring robust NO synthesis are not merely physiological goals but the critical requirements for preventing the structural remodelling associated with hypertension and atherosclerotic progression. Integration of these biological mechanisms is paramount for any rigorous pursuit of vascular longevity and systemic vitality.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Nitric Oxide and Endothelial Function: The Science of Vascular Elasticity"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper