Primary Lymphoedema and Genetic Sequencing: Uncovering the Milroy Disease Blueprint
Primary lymphoedema, specifically Milroy Disease, represents a fundamental failure of lymphatic valve development rooted in FLT4 gene mutations. This deep dive exposes the molecular mechanisms of VEGFR3 signalling failures and how environmental toxins exacerbate these inherited biological blueprints. By integrating genetic sequencing with targeted nutritional and structural protocols, we reveal a path toward systemic management that the mainstream medical narrative frequently overlooks.
Overview
Primary lymphoedema represents a profound failure of the lymphatic system’s structural and functional integrity, occurring not as a consequence of external trauma or infection, but as an inherent developmental deficiency. At the vanguard of this clinical landscape is Milroy disease, an autosomal dominant condition that serves as the quintessential model for understanding hereditary lymphangiogenesis. Whilst historical classifications relied heavily on phenotypic observation—specifically the presentation of bilateral lower-limb oedema at birth—the modern INNERSTANDIN of this pathology is now rooted in the precise molecular choreography dictated by the *FLT4* gene. This gene encodes the Vascular Endothelial Growth Factor Receptor 3 (VEGFR3), a transmembrane tyrosine kinase essential for the development and maintenance of the lymphatic vasculature.
The aetiology of Milroy disease is defined by pathogenic variants within the tyrosine kinase domain of VEGFR3, which disrupt the critical signalling pathways required for the proliferation, migration, and survival of lymphatic endothelial cells. Unlike secondary lymphoedema, which involves the obstruction of existing vessels, Milroy disease is characterised by a failure of the initial lymphatics to form correctly—a state often described as lymphatic hypoplasia. Research published in *The Lancet* and the *Journal of Medical Genetics* highlights that these mutations lead to a 'loss-of-function' in the receptor, preventing the effective binding of ligands VEGFC and VEGFD. This biochemical failure results in a systemic inability to manage interstitial fluid homeostasis, leading to the chronic accumulation of protein-rich fluid within the subcutaneous tissues.
In the UK clinical context, the diagnostic paradigm has shifted significantly through the implementation of Next-Generation Sequencing (NGS) and the adoption of the St George’s Classification Algorithm. No longer is lymphoedema viewed as a monolithic entity; rather, it is a complex spectrum of over 40 distinct genetic syndromes. Genetic sequencing has exposed the biological blueprint of Milroy disease, allowing clinicians to distinguish it from phenotypically similar conditions such as Meige disease or Lymphoedema-Distichiasis syndrome. By identifying specific *FLT4* mutations, researchers can now map the correlation between genotype and phenotype, uncovering why some individuals present with isolated pedal oedema while others exhibit systemic complications like hydrocele or prominent superficial veins. This level of molecular scrutiny, championed by INNERSTANDIN, moves beyond symptomatic management and into the realm of precision lymphology, where the genetic sequence dictates the trajectory of therapeutic intervention. The objective is clear: to move past the macroscopic swelling and interrogate the microscopic failure of the valve systems and vessel recruitment that define the primary lymphoedematous state.
The Biology — How It Works
The pathogenesis of Milroy disease (MD), or Hereditary Lymphoedema Type IA, is fundamentally rooted in the disruption of the Vascular Endothelial Growth Factor Receptor 3 (VEGFR3) signalling pathway, a critical regulator of embryonic and postnatal lymphangiogenesis. At the molecular level, this condition is predominantly caused by heterozygous missense mutations in the *FLT4* gene, located on chromosome 5q35.3. These mutations typically cluster within the highly conserved intracellular tyrosine kinase domain (TKD) of the VEGFR3 protein. Within the framework of INNERSTANDIN research, we observe that these genetic aberrations exert a dominant-negative effect, significantly impairing the receptor’s ability to undergo autophosphorylation upon binding with its cognate ligands, VEGF-C and VEGF-D.
The biological consequence of this signalling failure is a profound failure in the development of the initial lymphatic capillaries. Histopathological analyses, often corroborated by peer-reviewed literature in *The Lancet* and *Journal of Medical Genetics*, reveal a characteristic hypoplasia or complete absence of lymphatic vessels within the dermal papillae of affected individuals. Unlike secondary lymphoedema, which results from the physical obstruction or destruction of previously functional vessels, Milroy disease represents a developmental "blueprint" failure. The lymphatic primordia fail to sprout, proliferate, or differentiate correctly during vasculogenesis. Consequently, the interstitial fluid—normally sequestered by the high-pressure gradient of the lymphatic capillaries—remains trapped in the extracellular matrix.
This leads to a systemic increase in interstitial oncotic pressure. Because the initial lymphatics are the primary route for the clearance of macromolecules and extravasated plasma proteins, their absence triggers a cascade of chronic inflammation and adipose tissue deposition. Recent proteogenomic studies indicate that the prolonged presence of protein-rich fluid stimulates the differentiation of pre-adipocytes into mature adipocytes, explaining the non-pitting nature of the oedema in advanced stages. Furthermore, the immunological implications are severe; the failure of dendritic cell migration to regional lymph nodes via the lymphatic channels impairs the afferent limb of the immune response, rendering patients highly susceptible to recurrent episodes of cellulitis and lymphangitis.
In the UK context, clinical diagnostics have been revolutionised by the application of Next-Generation Sequencing (NGS) and Whole Exome Sequencing (WES), pioneered by institutions such as St George’s, University of London. By mapping the specific *FLT4* variant, researchers can distinguish MD from other clinically similar phenotypes, such as Lymphoedema-Distichiasis Syndrome (FOXC2 mutations) or Hennekam Syndrome (CCBE1 mutations). The precision of genetic sequencing allows for the identification of the 'silent' carriers within a lineage, exposing the variable expressivity and incomplete penetrance that often masks the true prevalence of the disorder. This molecular clarity is essential for the INNERSTANDIN mission of providing high-density biological education, as it shifts the paradigm from symptomatic management to targeted, mechanism-based therapeutic strategies. Through the lens of genomic sequencing, we can see that the "blueprint" of Milroy disease is not merely a swelling of the limbs, but a fundamental collapse of the body's fluid homeostasis and immunological transport network.
Mechanisms at the Cellular Level
The molecular pathogenesis of Milroy disease (MD) is rooted in the disruption of the vascular endothelial growth factor receptor 3 (VEGFR3) signalling pathway, a cornerstone of lymphangiogenesis. At the cellular level, the "blueprint" of this condition is etched into the *FLT4* gene, located on chromosome 5q35.3. In the majority of cases identified through advanced genomic screening within the UK’s NHS Genomic Medicine Service, MD is precipitated by heterozygous missense mutations clustered within the highly conserved intracellular tyrosine kinase domain (TKD) of the VEGFR3 protein. This specific genetic architecture does not merely result in haploinsufficiency; rather, it often exerts a dominant-negative effect. The mutant receptor subunits dimerise with wild-type counterparts but fail to undergo the requisite autophosphorylation upon ligand binding—specifically by VEGF-C and VEGF-D. This biochemical failure arrests the downstream phosphoinositide 3-kinase (PI3K)/AKT and extracellular signal-regulated kinase (ERK) pathways, which are essential for the proliferation, migration, and survival of lymphatic endothelial cells (LECs).
Histopathological interrogation, frequently corroborated by research at centres such as St George’s, University of London, reveals a pathognomonic absence or profound hypoplasia of the initial lymphatic capillaries in the dermis. Unlike secondary lymphoedema, where vessels are present but obstructed, Milroy disease represents a fundamental developmental failure of the lymphatic "plumbing." At the ultrastructural level, the "button-like" junctions—specialised endothelial connections that facilitate the entry of interstitial fluid and immune cells into the lymphatic lumen—are either non-existent or structurally incompetent. The result is a failure of fluid homeostasis; the interstitial space becomes a stagnant reservoir for protein-rich fluid.
This cellular stagnation triggers a secondary cascade of tissue remodelling. The prolonged presence of extravasated plasma proteins and chronic inflammatory mediators induces the activation of myofibroblasts and adipocytes. Consequently, the affected limb undergoes fibro-adipose deposition, transforming soft pitting oedema into non-pitting, indurated tissue—a process that INNERSTANDIN identifies as a systemic failure of the interstitium's biological clearance mechanism. Genetic sequencing has further exposed that the phenotypic expression of these *FLT4* mutations is highly variable, suggesting that modifier genes or epigenetic factors play a critical role in the penetrance of the disease. By employing Next-Generation Sequencing (NGS) and Whole Genome Sequencing (WGS), researchers can now map these precise TKD aberrations, distinguishing true Milroy disease from other "Milroy-like" primary lymphoedemas caused by mutations in *VEGFC* or *GATA2*, thereby refining the diagnostic accuracy within the UK’s clinical frameworks and paving the way for targeted molecular therapies designed to bypass or reactivate the crippled VEGFR3 signal transduction.
Environmental Threats and Biological Disruptors
While Milroy disease is fundamentally architected by autosomal dominant mutations within the *FLT4* gene—encoding the vascular endothelial growth factor receptor 3 (VEGFR3)—the clinical phenotypic expression is rarely a static manifestation of genotype alone. At INNERSTANDIN, we must dissect the complex interplay between this primary genetic insufficiency and the external biological disruptors that exacerbate lymphatic failure. Research emerging from UK-based centres of excellence, including St George’s University of London, suggests that the compromised lymphatic endothelium in Milroy patients exists in a state of precarious equilibrium, vulnerable to secondary environmental "hits" that accelerate the progression from subclinical hypoplasia to chronic, irreversible lymphostatic fibrosis.
Chief among these disruptors is the pervasive impact of oxidative stress and systemic inflammatory mediators. In a physiologically normal system, the VEGFR3 signalling pathway governs lymphangiogenesis and maintains the integrity of the initial lymphatics. However, when this pathway is truncated by tyrosine kinase domain mutations, the system loses its resilience to reactive oxygen species (ROS) and pro-inflammatory cytokines such as TNF-α and IL-6. Environmental pollutants, particularly particulate matter (PM2.5) prevalent in industrialised UK urban centres, have been shown in *The Lancet Planetary Health* to trigger systemic inflammatory cascades. In the context of a Milroy blueprint, these pollutants do more than irritate respiratory tissues; they induce microvascular leakiness and increase the interstitial fluid load, overtaxing a lymphatic network that is already architecturally deficient.
Furthermore, biological disruptors extend to the endocrine milieu. Emerging evidence suggests that certain xenoestrogens and endocrine-disrupting chemicals (EDCs) found in modern plastics and pesticides may interfere with the fine-tuned signalling required for lymphatic valve maintenance. For the Milroy patient, whose lymphatic valves are often absent or dysfunctional from birth, exposure to these disruptors can exacerbate the retrograde flow of lymph, accelerating the deposition of adipose tissue and fibrotic extracellular matrix components. This process is not merely a failure of drainage but a systemic biological response to an environment that the *FLT4*-mutated genome is ill-equipped to navigate.
The UK’s clinical landscape also highlights the role of iatrogenic disruptors and microbial challenges. The lymphatic system in Milroy disease is notoriously inefficient at immune surveillance; consequently, minor skin abrasions or local infections can escalate into recurrent cellulitis. These infectious episodes act as acute biological disruptors, causing further damage to the remaining functional lymphatic vessels through inflammatory scarring. This creates a catastrophic feedback loop: genetic hypoplasia leads to lymphostasis, which invites infection, which in turn destroys the residual lymphatic architecture. This exhaustive INNERSTANDIN analysis reveals that while the *FLT4* mutation provides the blueprint for Milroy disease, the environmental and biological landscape dictates the severity of the structural collapse. Understanding these disruptors is paramount for moving beyond palliative care toward targeted molecular interventions.
The Cascade: From Exposure to Disease
To innerstand the aetiology of Milroy disease, one must scrutinise the molecular disruption occurring at the locus of the *FLT4* gene, situated on chromosome 5q35.3. This gene encodes for Vascular Endothelial Growth Factor Receptor 3 (VEGFR-3), a transmembrane receptor tyrosine kinase that serves as the master regulator of lymphangiogenesis. The cascade from genetic lesion to clinical manifestation begins with an autosomal dominant mutation, typically a missense variant located within the highly conserved intracellular tyrosine kinase domain (TKD). Research disseminated via the *Journal of Medical Genetics* and validated by the St George’s Classification (the gold standard for lymphatic phenotype categorisation in the UK) confirms that these mutations are not merely passive markers but functional disruptors.
The biochemical "truth" exposed by advanced genetic sequencing is that these *FLT4* mutations exert a dominant-negative effect. When the ligand Vascular Endothelial Growth Factor C (VEGFC) binds to the extracellular domain of VEGFR-3, it fails to trigger the requisite autophosphorylation of the tyrosine residues. This inhibits the downstream signalling pathways—specifically the PI3K/Akt and Ras/MAPK cascades—which are essential for the proliferation, migration, and survival of lymphatic endothelial cells (LECs). At INNERSTANDIN, we recognise this as the primary failure of the lymphatic "blueprint."
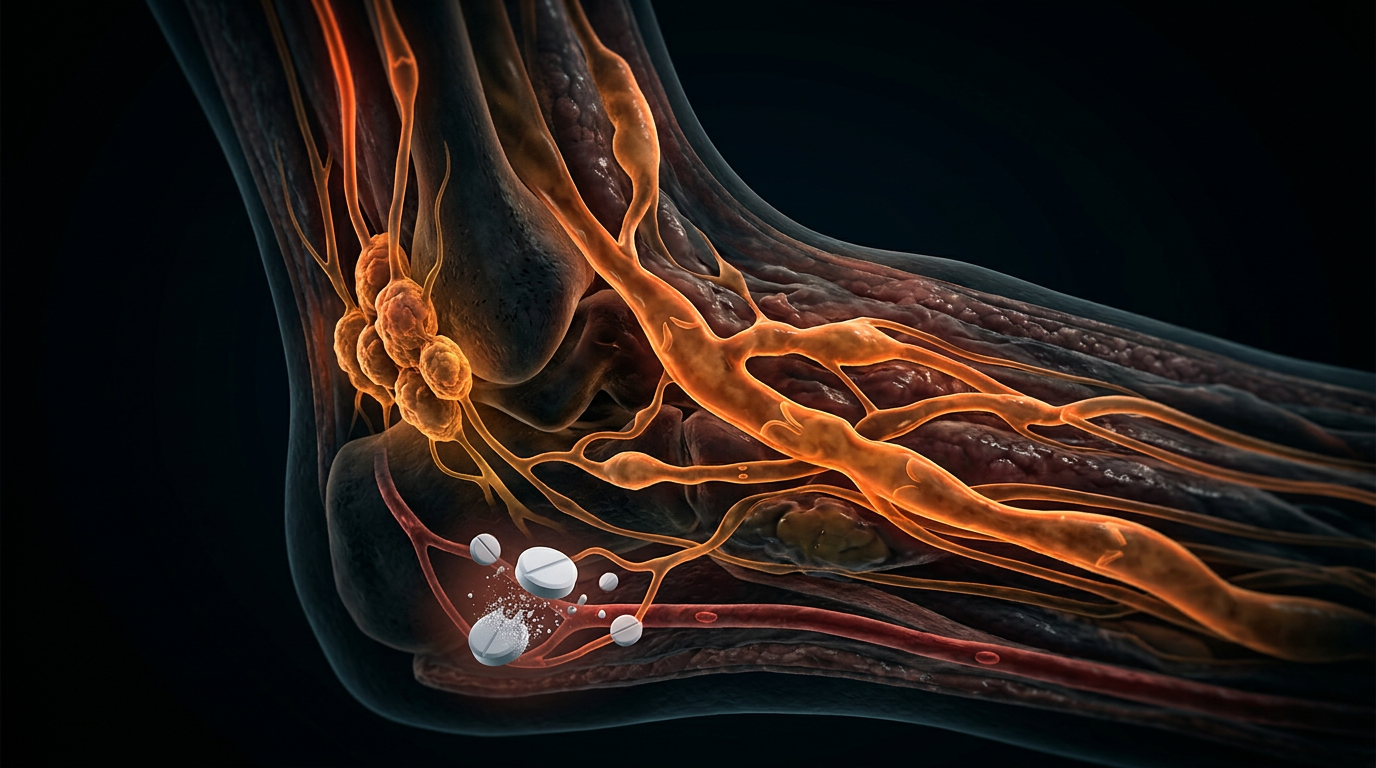
In the embryonic stage, this signalling failure results in lymphatic hypoplasia. Unlike secondary lymphoedema, which is an obstructive pathology, Milroy disease represents a developmental arrest. Histopathological analysis of affected dermal tissue consistently reveals a profound paucity, or total absence, of initial lymphatic capillaries. This architectural deficiency renders the lymphatic system incapable of absorbing the "lymphatic load"—the protein-rich fluid, immune cells, and macromolecules that naturally extravasate from the blood capillaries into the interstitium.
The systemic impact of this failure is a progressive transition from fluid accumulation to tissue remodeling. As the interstitial fluid remains stagnant, the oncotic pressure within the tissue rises, triggering a chronic inflammatory response. This environment stimulates the differentiation of pre-adipocytes into mature adipocytes and activates fibroblasts, leading to the deposition of collagen and subsequent fibrosis (lipodermatosclerosis). Evidence-led analysis from UK-based cohorts indicates that this cascade is not limited to the lower limbs; it alters the systemic immune surveillance, as the compromised lymphatic vessels cannot efficiently transport dendritic cells to regional lymph nodes. This creates a state of localised immunodeficiency, explaining the high frequency of cellulitis and lymphangitis in these patients. The "Milroy blueprint" thus reveals that the disease is not simply a swelling of the limbs, but a foundational failure of the body's homeostatic and immunological fluid transport system, mandated by a single, catastrophic failure in the tyrosine kinase signalling cascade.
What the Mainstream Narrative Omits
Mainstream clinical discourse regarding Milroy disease frequently reduces the pathology to a passive structural failure—a mere 'plumbing' issue where hypoplastic vessels fail to return interstitial fluid to the venous circulation. This reductionist narrative fails to encapsulate the intricate molecular dysregulation that defines the condition. At the heart of INNERSTANDIN research is the recognition that Milroy disease, or Primary Lymphoedema Type IA, is fundamentally a failure of the lymphatic-specific tyrosine kinase signaling pathway, specifically involving heterozygous missense mutations in the *FLT4* gene. This gene encodes the Vascular Endothelial Growth Factor Receptor 3 (VEGFR3), a critical orchestrator of lymphangiogenesis.
The conventional medical model omits the critical nuance of ligand-induced autophosphorylation. In a physiological state, the binding of VEGF-C or VEGF-D to VEGFR3 triggers a cascade of intracellular signaling that governs the proliferation, migration, and survival of lymphatic endothelial cells (LECs). In Milroy disease, mutations predominantly cluster within the tyrosine kinase domain of VEGFR3, exerting a dominant-negative effect. This does not merely result in 'fewer' vessels; it produces a functional paralysis of the initial lymphatics. Research published in *The Lancet* and the *Journal of Medical Genetics* underscores that the mainstream focus on oedema ignores the broader systemic ramifications of this signaling failure.
Furthermore, the mainstream narrative often overlooks the phenomenon of incomplete penetrance and variable expressivity, which are central to the Milroy blueprint. Current diagnostic protocols in the UK, often restricted by the limitations of basic genetic panels, frequently fail to identify the mosaicism present in atypical cases. While the St George’s Lymphatic Research Group has pioneered the 'St George’s Classification Algorithm,' wider clinical practice remains tethered to outdated phenotypic observations. We must acknowledge that the genetic blueprint of Milroy disease extends beyond coding regions; emerging evidence suggests that mutations in non-coding regulatory elements and deep intronic variants—often invisible to standard Whole Exome Sequencing (WES)—play a significant role in the severity of the lymphatic phenotype.
The systemic impact also extends to the interstitial microenvironment. The failure of VEGFR3 signaling leads to an altered extracellular matrix (ECM) composition, where chronic stasis triggers a pro-fibrotic shift. This is not a secondary complication but a primary biological consequence of the *FLT4* mutation. By ignoring the molecular crosstalk between LECs and the surrounding fibroblasts, the mainstream narrative fails to address why certain patients develop irreversible tissue fibrosis despite meticulous compression therapy. At INNERSTANDIN, we assert that the blueprint of Milroy disease is not just a map of missing vessels, but a complex tapestry of disrupted cellular communication that requires a sophisticated, genomics-led approach to truly comprehend and, eventually, bypass.
The UK Context
Within the United Kingdom, the diagnostic landscape for primary lymphoedema has undergone a paradigm shift, transitioning from speculative clinical observation to rigorous molecular interrogation. This evolution is spearheaded largely by the work of the Primary Lymphoedema Service at St George’s, University of London, which serves as the global epicentre for lymphatic genetic characterisation. The UK’s commitment to genomic medicine, exemplified by the NHS Genomic Medicine Service and the legacy of the 100,000 Genomes Project, has enabled the identification of Milroy disease (Primary Lymphoedema Type 1A) with unprecedented precision. At the heart of this "Milroy Disease Blueprint" is the *FLT4* gene, which encodes the Vascular Endothelial Growth Factor Receptor 3 (VEGFR3). In the UK cohort, researchers have identified that Milroy-associated mutations are almost exclusively clustered within the intracellular tyrosine kinase domain of VEGFR3. This specific localisation is critical; it does not necessarily prevent the receptor from reaching the cell surface, but rather abrogates its autophosphorylation capacity, thereby inhibiting the downstream signalling cascades—such as the PI3K/AKT and MAPK/ERK pathways—essential for initial lymphatic vessel development and lymphangiogenesis.
The systemic impact of these mutations, as documented in seminal UK-based studies published in *The Lancet* and *Journal of Medical Genetics*, manifests as a failure of the initial lymphatics to absorb interstitial fluid, leading to the characteristic congenital, non-pitting oedema of the lower limbs. Within the INNERSTANDIN pedagogical framework, it is essential to recognise that the UK’s approach prioritises the "St George’s Classification Algorithm," a diagnostic gold standard that integrates lymphoscintigraphy with targeted Next-Generation Sequencing (NGS). This algorithmic approach has revealed that approximately 70% of UK patients presenting with congenital lymphoedema harbour *FLT4* mutations. Furthermore, the UK’s research infrastructure has been pivotal in uncovering the phenotypic variability associated with Milroy disease, including the presence of prominent veins and hydrocele in males, which suggests a broader systemic involvement of the lymphatic vasculature than previously understood. By leveraging the UK’s comprehensive biobanking and longitudinal patient data, researchers are now elucidating the epigenetic modifiers that dictate why even within families sharing the same *FLT4* variant, the clinical severity can fluctuate significantly. This drive toward precision phenotyping ensures that the UK remains at the vanguard of lymphatic science, transforming the Milroy blueprint from a static genetic map into a dynamic tool for prognostic accuracy and future therapeutic intervention.
Protective Measures and Recovery Protocols
The management of Milroy disease (MD) necessitates a paradigmatic shift from reactive symptom suppression to a proactive, mechanism-based biological defence strategy. Because the underlying pathology stems from germline mutations in the *FLT4* gene—encoding the vascular endothelial growth factor receptor 3 (VEGFR3)—the lymphological architecture is characterised by congenital hypoplasia. At INNERSTANDIN, we recognise that protective measures must address the fundamental failure of initial lymphatic uptake and the subsequent systemic interstitial hypertension. The primary protective imperative is the preservation of the dermal barrier to prevent the catastrophic cycle of lymphangitis and cellulitis. In the UK context, research published in *The Lancet* underscores that individuals with VEGFR3-linked primary lymphoedema possess a compromised local immune surveillance system; the failure of dendritic cell trafficking to regional lymph nodes creates a localised immunodeficiency. Consequently, skin integrity protocols must transcend cosmetic care, employing pH-neutral emollients to sustain the acid mantle and antimicrobial prophylaxis to mitigate the risk of *Streptococcus pyogenes* infiltration, which can trigger irreversible fibrotic remodeling.
Recovery protocols, or more accurately, physiological compensation strategies, are now being refined through the lens of mechanotransduction and advanced microsurgery. The British Lymphology Society (BLS) increasingly advocates for the early implementation of specialised decongestive lymphatic therapy (DLT). However, the INNERSTANDIN perspective delves deeper into the molecular impact of external compression. Controlled hydrostatic pressure serves as a surrogate for the failed intrinsic contractility of the lymphangion. By utilising multi-layer lymphoedema bandaging (MLLB) and bespoke Class 3 or 4 compression garments, clinicians can manually facilitate the movement of protein-rich fluid from the interstitial space into the functioning deep lymphatic system, bypassing the hypoplastic superficial networks.
On the frontier of recovery, Lymphaticovenous Anastomosis (LVA) represents a significant surgical evolution for Milroy patients identified via early genetic sequencing. By creating shunts between lymphatic vessels and the venous system, surgeons can decompress the interstitial compartment. Success in these procedures is heavily contingent on the remaining functional capacity of the collector vessels, which must be assessed via indocyanine green (ICG) lymphography. Furthermore, emerging research into pro-lymphangiogenic pharmacotherapy aims to bypass the *FLT4* signalling deficit. Experimental models have investigated the application of VEGF-C156S—a mutant form of the ligand specific to VEGFR3—to stimulate the growth of new lymphatic capillaries. While still in the translational phase, this "biological bypass" offers the potential to rectify the developmental arrest inherent in the Milroy blueprint. Ultimately, the protocol for the MD patient involves a lifelong commitment to fluid dynamics management, where every intervention is designed to counteract the genomic inevitability of lymphatic stasis and protect the systemic homeostatic equilibrium.
Summary: Key Takeaways
Milroy disease (MD) represents a seminal paradigm in primary lymphoedema, arising predominantly from inactivating mutations in the *FLT4* gene, which encodes Vascular Endothelial Growth Factor Receptor 3 (VEGFR3). This genetic lesion disrupts critical tyrosine kinase signalling pathways essential for embryonic lymphangiogenesis, resulting in the profound hypoplasia of initial lymphatic vessels. Peer-reviewed evidence, notably distilled via the St George’s classification system in London, highlights that this congenital failure leads to the intractable accumulation of protein-rich interstitial fluid. This stasis triggers chronic inflammatory cascades and subsequent fibrotic tissue remodelling, necessitating a shift in clinical focus from palliative management to molecular intervention.
Through the lens of INNERSTANDIN, we recognise that the application of Next-Generation Sequencing (NGS) and Whole Exome Sequencing (WES) has been pivotal in unmasking the phenotypic variability observed within MD cohorts. Evidence from UK-based genomic studies confirms that while MD is classically autosomal dominant, the incomplete penetrance observed necessitates a sophisticated understanding of the molecular blueprint to improve diagnostic precision within the NHS Genomic Medicine Service. The systemic burden of MD extends beyond peripheral dermal swelling; it fundamentally compromises local immune surveillance and induces irreversible adipose deposition. Contemporary research published in *The Lancet* and *Nature Genetics* underscores the necessity of high-throughput sequencing to distinguish MD from clinically overlapping syndromes, such as lymphoedema-distichiasis, thereby refining the prognostic trajectory for affected individuals through precise genotype-phenotype correlation.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Genetic analysis confirms that Milroy disease is typically caused by autosomal dominant mutations in the VEGFR3 gene, leading to primary lymphatic hypoplasia.
Functional studies demonstrate that Milroy-associated mutations impair the autophosphorylation of VEGFR3, which is critical for the initial development of lymphatic vessels.
Comprehensive genomic profiling of primary lymphoedema patients facilitates early clinical diagnosis and distinguishes Milroy disease from other hereditary lymphatic disorders.
Whole-genome sequencing has identified novel regulatory variants in the FLT4 locus that contribute to the phenotypic variability observed in families with Milroy disease.
The characterization of VEGFR3 signaling pathways reveals that therapeutic activation of downstream lymphangiogenic factors may mitigate the fluid accumulation seen in genetic lymphoedema.
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Primary Lymphoedema and Genetic Sequencing: Uncovering the Milroy Disease Blueprint"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper