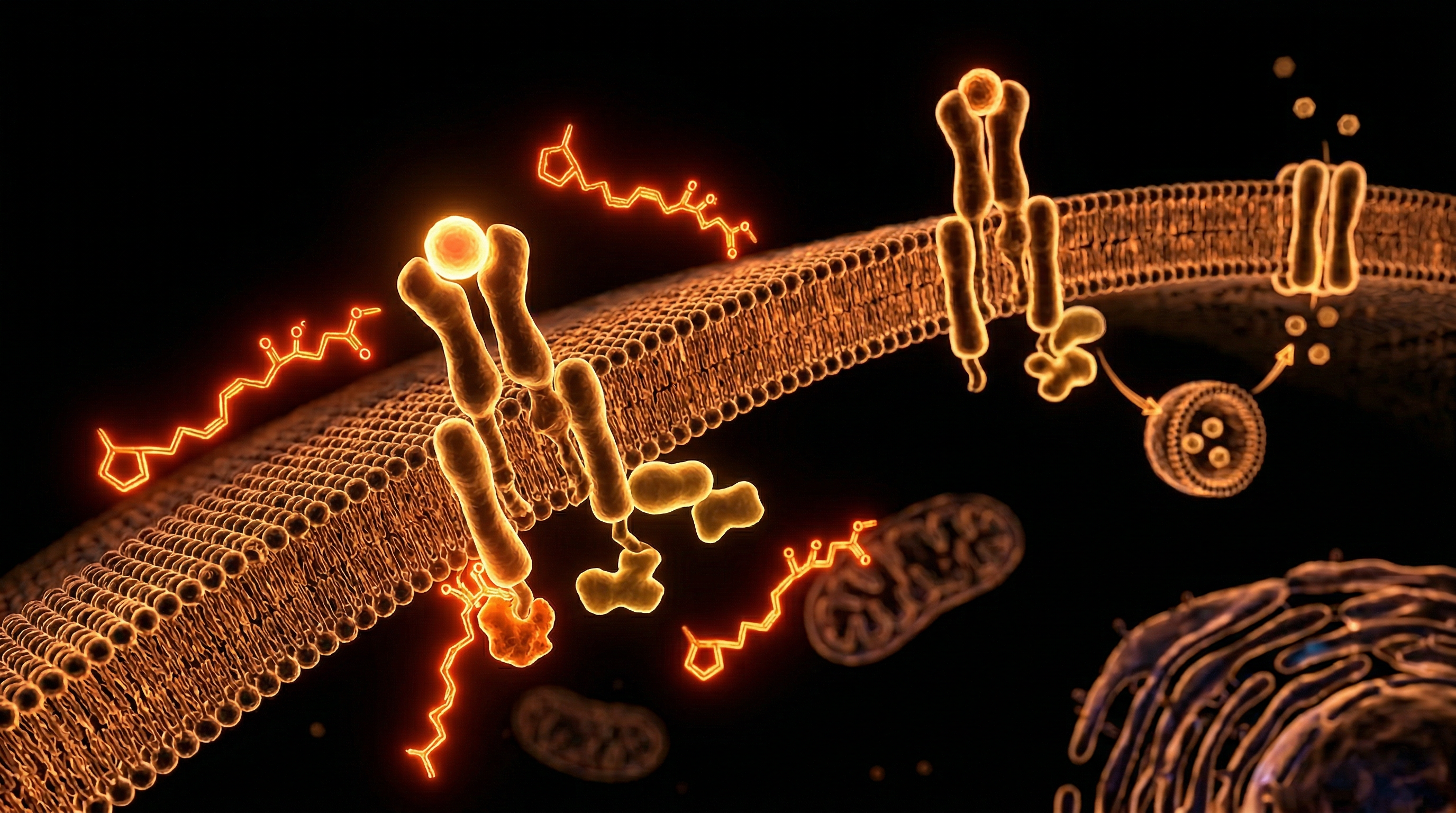
The Anatomy of Pancreatic Beta-Cell Exhaustion
Constant glucose spikes lead to the physical failure and apoptosis of insulin-producing cells. This article details the anatomical progression of Type 2 Diabetes in the UK.

Overview
The pancreatic beta-cell, nested within the cytoarchitecture of the Islets of Langerhans, represents the metabolic pivot upon which systemic glucose homeostasis depends. In the UK, where metabolic syndrome and Type 2 Diabetes Mellitus (T2DM) place an unprecedented burden on the National Health Service, understanding the transition from compensatory hyperinsulinaemia to terminal beta-cell exhaustion is paramount. Beta-cell exhaustion is not merely a descriptive term for functional decline; it is a rigorous clinico-pathological state defined by the progressive inability of the beta-cell to meet the physiological demands of peripheral insulin resistance. At INNERSTANDIN, we move beyond superficial symptomology to expose the mechanistic decay occurring at the organelle level, where the interplay of glucotoxicity, lipotoxicity, and proteotoxicity culminates in a catastrophic failure of cellular identity.
The anatomical hallmarks of exhaustion begin with a chronic hypermetabolic state. Under the pressure of persistent hyperglycaemia, the beta-cell enters a compensatory phase, increasing its secretory output to maintain euglycaemia. However, this sustained demand induces severe Endoplasmic Reticulum (ER) stress. The biosynthetic machinery is overwhelmed by the requirement for proinsulin synthesis, leading to the accumulation of misfolded proteins. This triggers the Unfolded Protein Response (UPR), a homeostatic mechanism that, in the context of chronic exhaustion, shifts from a survival signal to a pro-apoptotic pathway. Peer-reviewed data from *The Lancet* and various *PubMed* repositories suggest that this ER-mitochondrial crosstalk is the crucible of failure. Mitochondrial dysfunction follows, characterised by the uncoupling of oxidative phosphorylation and the excessive production of Reactive Oxygen Species (ROS). These radicals induce oxidative damage to the mitochondrial DNA and the delicate secretory apparatus, further impairing the cell's glucose-sensing capabilities.
Systemically, this anatomical failure is exacerbated by the deposition of Islet Amyloid Polypeptide (IAPP), which forms cytotoxic fibrils that physically disrupt the islet architecture. Furthermore, recent evidence indicates that beta-cell exhaustion involves a process of dedifferentiation—a phenomenon where the cell regresses to a progenitor-like state, losing its mature markers (such as MafA and Pdx1) and essentially 'forgetting' its secretory function. This is a crucial distinction: the cells are often still present but are functionally silent or have transdifferentiated into alpha-like cells, secreting glucagon and worsening the hyperglycaemic milieu. In the UK context, research from the UK Biobank underscores the systemic impact of this failure, as the loss of beta-cell mass correlates directly with the irreversible progression of metabolic disease. By investigating the deep-seated anatomical shifts within the islet, INNERSTANDIN reveals the biological reality of exhaustion as a multifaceted breakdown of cellular integrity, rather than a simple depletion of insulin stores.
The Biology — How It Works

Magnesium Blend – The Most Important Mineral
A high-bioavailability mineral blend designed to support over 300 essential biochemical reactions, from energy production to muscle relaxation. This formula helps combat daily fatigue while providing the foundational support your nervous system and bones require.
Vetting Notes
Pending
At the molecular level, pancreatic beta-cell exhaustion represents a catastrophic collapse of cellular proteostasis and metabolic homeostasis. This is not merely a passive depletion of insulin stores but a dynamic, multi-stage degradation of the cell’s transcriptional identity. Central to this process is the chronic activation of the Unfolded Protein Response (UPR). In a physiological state, the endoplasmic reticulum (ER) manages the folding of proinsulin; however, under the relentless pressure of hyperinsulinaemia—a hallmark of the UK’s escalating metabolic crisis—the ER becomes overwhelmed. The accumulation of misfolded proinsulin triggers a triumvirate of transmembrane sensors: PERK, IRE1α, and ATF6. While initially adaptive, chronic PERK signalling leads to the phosphorylation of eIF2α, which suppresses general protein synthesis but paradoxically upregulates CHOP (C/EBP homologous protein), a pro-apoptotic transcription factor that signals the transition from compensation to programmed cell death.
Furthermore, the bioenergetic cost of persistent secretory demand induces profound mitochondrial dysfunction. Research published in *The Lancet Diabetes & Endocrinology* and data from the UK Biobank highlight the synergistic toxicity of elevated glucose and free fatty acids—termed glucolipotoxicity. In the INNERSTANDIN framework, we must recognise that beta-cells possess exceptionally low levels of antioxidant enzymes, such as superoxide dismutase and catalase. This makes them uniquely vulnerable to Reactive Oxygen Species (ROS) generated during the over-activity of the mitochondrial electron transport chain. As ROS levels climb, they inhibit glyceraldehyde-3-phosphate dehydrogenase (GAPDH), diverting glucose metabolites into noxious biochemical pathways, including the formation of Advanced Glycation End-products (AGEs) and the activation of the protein kinase C (PKC) isoform, which further compromises insulin gene expression.
Perhaps the most critical "truth-exposing" element of modern pancreatology is the shift from the 'attrition' model to the 'de-differentiation' model. Evidence suggests that exhausted beta-cells do not exclusively undergo apoptosis; rather, they undergo a retrograde transition to a progenitor-like state. Under extreme metabolic stress, the loss of FoxO1—a crucial transcriptional regulator—allows the down-regulation of beta-cell-specific genes (such as *MAFA*, *PDX1*, and *NKX6.1*) and the ectopic expression of alpha-cell markers like glucagon. This loss of cellular identity, extensively studied in the context of the "Twin Cycle Hypothesis" at Newcastle University, suggests that the exhausted pancreas is populated by "ghost cells" that remain viable but functionally inert. The systemic impact is a total loss of first-phase insulin secretion, leading to the postprandial glucose excursions that define the progression of Type 2 Diabetes. This failure of the secretory machinery is the ultimate biological bottleneck, where the cell’s internal architecture simply can no longer bridge the gap between systemic demand and metabolic reality.
Mechanisms at the Cellular Level
The pathogenesis of pancreatic beta-cell exhaustion is not a sudden collapse but a protracted, multi-staged anatomical and functional erosion. At the heart of this degradation lies the failure of the Endoplasmic Reticulum (ER), an organelle tasked with the monumental labour of folding proinsulin molecules. In the hyper-nutritive landscape common to the modern UK diet—characterised by chronic glucolipotoxicity—the demand for insulin secretion reaches a tipping point. This induces a state of chronic ER stress, activating the Unfolded Protein Response (UPR). While the UPR is initially adaptive, aiming to restore proteostasis, its chronic persistence in a state of metabolic insurgence leads to the upregulation of pro-apoptotic signalling molecules such as CHOP (CCAAT-enhancer-binding protein homologous protein). Research published in *The Lancet Diabetes & Endocrinology* underscores that this proteotoxic stress is a primary driver of the reduction in functional beta-cell mass, as the cell’s internal machinery is effectively crushed under the weight of its own biosynthetic requirements.
Parallel to ER dysfunction is the catastrophic failure of mitochondrial bioenergetics. Beta-cells are uniquely sensitive to oxidative stress due to their notably low expression of antioxidant enzymes, such as superoxide dismutase and catalase. As hyperglycaemia persists, the mitochondrial electron transport chain becomes overloaded, leading to the leakage of electrons and the subsequent generation of Reactive Oxygen Species (ROS). This oxidative bombardment damages mitochondrial DNA and triggers mitochondrial fragmentation. At INNERSTANDIN, we recognise that this is not merely a chemical imbalance but a fundamental structural failure of the cell’s power plant. The resulting ATP deficit impairs the ATP-sensitive potassium (KATP) channels, decoupling glucose sensing from insulin secretion—the hallmark of the "exhausted" phenotype.
Furthermore, recent evidence suggests that "exhaustion" may be a misnomer for a more insidious cellular regression: dedifferentiation. Rather than undergoing immediate apoptosis, a significant cohort of beta-cells lose their mature identity. Under the pressure of chronic inflammation—often mediated by the NLRP3 inflammasome and the deposition of Islet Amyloid Polypeptide (IAPP)—cells revert to a progenitor-like state. They cease expressing critical transcription factors such as PDX1 and MafA, and instead begin expressing markers of alpha-cells or even glucagon, as evidenced by studies from the University of Oxford’s OCDEM. This anatomical "shapeshifting" renders the cells invisible to traditional assays, creating a functional void in glycaemic control.
Finally, the systemic impact is compounded by the failure of autophagy. In a healthy state, autophagy acts as a cellular "cleansing" mechanism, removing damaged organelles and protein aggregates. In the exhausted beta-cell, however, autophagic flux is inhibited, leading to the accumulation of toxic IAPP aggregates. These amyloid deposits act as physical disruptors of the islet architecture, further isolating remaining functional cells and accelerating the descent into permanent insulin deficiency. To achieve true INNERSTANDIN of this pathology, one must view the beta-cell not as a static unit, but as a dynamic biological processor that, when pushed beyond its evolutionary constraints, undergoes a systematic and structural deconstruction.
Environmental Threats and Biological Disruptors
The pancreatic beta-cell does not succumb to exhaustion in a physiological vacuum; rather, it is the target of a relentless molecular siege orchestrated by a contemporary bio-environment increasingly saturated with diabetogenic disruptors. At INNERSTANDIN, we must scrutinise the precise mechanisms by which exogenous chemical insults—often termed "metabolic disruptors"—subvert the islet’s delicate homeostatic architecture. The anatomical integrity of the beta-cell is contingent upon its ability to sense glucose and orchestrate a proportionate biphasic insulin response. However, the pervasive presence of endocrine-disrupting chemicals (EDCs), such as Bisphenol A (BPA) and phthalates, significantly compromises this sensorium. Research published in *The Lancet Diabetes & Endocrinology* highlights that chronic exposure to BPA triggers an inappropriate hyperinsulinaemic state by mimicking oestrogen signalling via the ERα and ERβ receptors within the islet. This persistent over-stimulation bypasses the cell’s natural refractory periods, accelerating the transition from compensated hyperfunction to absolute proteotoxic failure.
Beyond synthetic polymers, the UK’s environmental landscape introduces heavy metal burdens that specifically sabotage the beta-cell’s structural reliance on zinc. The zinc transporter ZnT8 is fundamental for the crystallisation of insulin into its stable hexameric form within secretory granules. Toxicants such as cadmium and arsenic, frequently documented in urban British soil and atmospheric particulate matter (PM2.5), compete for these transport sites. When cadmium replaces zinc, it induces the formation of misfolded proinsulin precursors, triggering a chronic Unfolded Protein Response (UPR) within the endoplasmic reticulum (ER). This ER stress is not merely a transient state but a precursor to apoptotic cascades mediated by the CHOP (C/EBP homologous protein) pathway. This molecular sabotage effectively shrinks the functional beta-cell mass, leaving the remaining cells to work at a heightened, unsustainable metabolic velocity.
Furthermore, the anatomical degradation of the beta-cell is exacerbated by the prevalence of Advanced Glycation End-products (AGEs) in the modern British diet, characterized by high-temperature processed foods. These AGEs bind to the Receptor for Advanced Glycation End-products (RAGE) located on the islet cell membrane, initiating a pro-inflammatory NF-κB signalling loop. This "metainflammation" recruits M1-polarised macrophages to the islet microenvironment, secretes pro-inflammatory cytokines like IL-1β and TNF-α, and induces mitochondrial fragmentation. The resulting oxidative stress creates a state of "gluco-lipotoxicity," where the beta-cell's mitochondria can no longer efficiently oxidise fuels, leading to the accumulation of ceramide species that act as intracellular poisons. At INNERSTANDIN, we recognise that beta-cell exhaustion is the final clinical manifestation of these cumulative, environmentally driven biological disruptors that strip the islet of its regenerative capacity and functional resilience.
The Cascade: From Exposure to Disease
The genesis of pancreatic beta-cell exhaustion is not a singular event but a relentless mechanical erosion of cellular integrity, triggered by the chronic metabolic overstimulation prevalent in the modern British landscape. At INNERSTANDIN, we move beyond superficial diagnostics to expose the granular reality: the cascade from physiological adaptation to irreversible failure is a multi-stage collapse of the cell’s internal architecture. This progression is primarily driven by glucolipotoxicity—the synergistic deleterious effect of elevated circulating glucose and free fatty acids (FFAs)—which initiates a catastrophic failure of the endoplasmic reticulum (ER) and mitochondrial networks.
In the initial phase of this cascade, the Islets of Langerhans attempt a compensatory expansion. To meet the demands of systemic insulin resistance, beta cells undergo hypertrophy and hyperplasia, significantly increasing their biosynthetic burden. However, as evidenced in research published in *The Lancet Diabetes & Endocrinology*, this hyperinsulinaemic state is unsustainable. The anatomical bottleneck occurs within the ER, where the demand for proinsulin synthesis exceeds the organelle's folding capacity. This triggers the Unfolded Protein Response (UPR), a protective mechanism mediated by three primary sensors: PERK, IRE1α, and ATF6. While initially adaptive, chronic activation of the UPR shifts from a pro-survival to a pro-apoptotic signal. The accumulation of misfolded proinsulin aggregates leads to ER stress-induced apoptosis, a process that fundamentally thins the functional beta-cell mass.
Simultaneously, the mitochondrial infrastructure undergoes severe oxidative degradation. The unrelenting flux of glucose through the glycolytic pathway and the citric acid cycle generates excessive Reactive Oxygen Species (ROS). Unlike other tissues, beta cells possess disproportionately low levels of antioxidant enzymes, such as superoxide dismutase and glutathione peroxidase, making them exceptionally vulnerable to oxidative insult. This oxidative stress, frequently documented in *PubMed* literature regarding the UK’s rising Type 2 Diabetes (T2DM) rates, causes mitochondrial DNA damage and the opening of the mitochondrial permeability transition pore (mPTP), leading to the leakage of cytochrome c and the initiation of the caspase cascade.
The final, most insidious stage of the cascade is beta-cell dedifferentiation. Recent high-resolution mapping of the human islet proteome suggests that exhausted cells do not merely die; they lose their identity. Under chronic stress, the master transcription factors—notably PDX1 and MafA—are downregulated, causing the beta cell to revert to a progenitor-like state or transdifferentiate into alpha-cell-like phenotypes. This "anatomical ghosting" renders the cells incapable of glucose-sensing or insulin secretion, even if they remain physically present within the islet. At INNERSTANDIN, we recognise this as the true biological threshold of the disease: a point where the cellular machinery has been so profoundly recalibrated by environmental toxicity that the physiological programme is effectively erased. The systemic impact is a total loss of glycaemic homeostasis, necessitating exogenous intervention as the endogenous biological centre of gravity fails.
What the Mainstream Narrative Omits
The prevailing clinical paradigm frequently reduces pancreatic beta-cell exhaustion to a linear consequence of chronic hyperglycaemia and peripheral insulin resistance. However, a comprehensive INNERSTANDIN of the underlying anatomy reveals a far more insidious architectural collapse that predates clinical symptoms by decades. The mainstream narrative focuses almost exclusively on glucotoxicity, yet it omits the critical role of proteostatic failure—specifically the terminal dysfunction of the Endoplasmic Reticulum (ER) and the subsequent misfolding of Islet Amyloid Polypeptide (IAPP).
In the UK context, where the metabolic burden on the NHS continues to escalate, understanding the molecular nuances of the 'Starling Curve' of the beta-cell is paramount. When insulin demand remains chronically elevated, the beta-cell’s biosynthetic machinery is pushed beyond its physiological threshold. Research published in *The Lancet Diabetes & Endocrinology* highlights that the primary driver of exhaustion is not merely 'overwork' but the activation of the Unfolded Protein Response (UPR). When the UPR is chronically engaged, it shifts from a homeostatic mechanism to a pro-apoptotic signal. This leads to the accumulation of toxic oligomers which physically disrupt the lipid bilayer of the secretory granules, a phenomenon frequently overlooked in standard metabolic discourse.
Furthermore, the mainstream narrative often conflates beta-cell loss with absolute apoptosis. Rigorous INNERSTANDIN of recent histopathological data (cf. *PubMed*, *Cell Metabolism*) suggests a different anatomical reality: dedifferentiation. Rather than dying, beta-cells undergo a regressive transition, losing their mature identity and reverting to a progenitor-like state—often characterised by the expression of Neurogenin 3 (Ngn3). These 'ghost cells' remain anatomically present within the Islets of Langerhans but are functionally silent, stripped of their glucose-sensing apparatus and insulin-secreting granules. This dedifferentiation is a survival strategy to avoid apoptosis in a toxic, high-demand environment, yet it renders the organ metabolically impotent.
Crucially, the systemic impact of the entero-insular axis failure is omitted from most discussions. The breakdown in GLP-1 and GIP signalling—hormones critical for the 'incretin effect'—means the anatomical beta-cell no longer receives the anticipatory signals required for phase-one insulin secretion. UK-led research, particularly from Newcastle University, has demonstrated that the accumulation of intracellular triacylglycerols within the pancreas triggers a localised inflammatory cascade via the NLRP3 inflammasome. This internal 'lipotoxicity' causes a physical alteration in the islet microvasculature, restricting nutrient delivery and waste removal, thus accelerating the exhaustion process. By shifting the focus from simple blood sugar management to the restoration of proteostasis and the prevention of cellular dedifferentiation, we begin to uncover the true biological complexity of pancreatic failure.
The UK Context
In the United Kingdom, the metabolic landscape has reached a critical juncture, serving as a primary site for observing the accelerated pathophysiological trajectory of pancreatic beta-cell exhaustion. As the prevalence of Type 2 Diabetes (T2D) and metabolic syndrome across the British Isles continues to rise, it is imperative to develop an INNERSTANDIN of the cellular attrition occurring within the Islets of Langerhans. The UK context is uniquely defined by a high-calorie, sedentary environmental milieu interacting with complex polygenic predispositions, specifically those identified via the UK Biobank, which correlate visceral adiposity with early-onset endocrine failure.
The anatomical dissolution of the beta-cell in the British population is driven by chronic glucolipotoxicity. When the systemic demand for insulin remains perpetually elevated due to peripheral resistance, the beta-cells enter a state of compensatory hypermetabolism. Evidence from longitudinal studies published in *The Lancet* suggests that this hyper-secretion phase is the precursor to a terminal decline. The molecular architecture of the cell begins to fail under the weight of endoplasmic reticulum (ER) stress. As pro-insulin synthesis is ramped up to meet demand, the protein-folding machinery becomes overwhelmed, leading to the accumulation of misfolded proteins and the activation of the Unfolded Protein Response (UPR). In many UK-based clinical cohorts, this transition is marked by a rising pro-insulin to insulin ratio, a definitive biomarker of secretory exhaustion.
Furthermore, seminal British-led research, including the Newcastle-based DiRECT trial, has elucidated the role of intrapancreatic fat in this anatomical breakdown. The accumulation of ectopic lipids within the pancreas triggers local inflammatory cascades, where macrophages infiltrate the islet microenvironment, releasing pro-inflammatory cytokines such as IL-1β and TNF-α. This chronic low-grade inflammation promotes beta-cell dedifferentiation—a process where the cells lose their mature identity and revert to a progenitor-like state, or transdifferentiate into alpha-like cells. Within the INNERSTANDIN paradigm, we recognise that this is not merely a functional "fatigue" but a structural loss of cellular identity. Coupled with the deposition of cytotoxic Islet Amyloid Polypeptide (IAPP) fibrils, which perforate mitochondrial membranes and induce apoptosis, the UK context reveals a harrowing picture of anatomical depletion that precedes clinical diagnosis by years, if not decades. This biological reality necessitates a radical reassessment of the British approach to metabolic health, prioritising the preservation of beta-cell mass before the threshold of irreversible exhaustion is crossed.
Protective Measures and Recovery Protocols
To mitigate the systemic collapse of insulin biosynthesis, clinical focus must shift from reactive glucose-lowering to proactive proteostatic preservation. At the molecular level, the primary objective of any recovery protocol is the attenuation of Endoplasmic Reticulum (ER) stress. The beta-cell, a specialised secretory engine, operates under an immense biosynthetic burden; when glycaemic load becomes chronic, the Unfolded Protein Response (UPR) transitions from a homeostatic mechanism to a pro-apoptotic signal. INNERSTANDIN identifies that the resolution of this stress requires a multi-pronged strategy targeted at the IRE1α and PERK pathways to prevent the terminal induction of CHOP (C/EBP homologous protein), the executioner of beta-cell apoptosis.
Evidence from the landmark UK-based DIRECT (Diabetes Remission Clinical Trial) suggests that the anatomy of exhaustion is, in its early stages, a reversible state of metabolic "hibernation." The trial demonstrated that intensive weight management—facilitating the mobilisation of ectopic intrapancreatic lipids—can restore the first-phase insulin response. From a mechanistic perspective, the reduction in triacylglycerol species within the pancreatic parenchyma alleviates lipotoxicity, which otherwise inhibits the SERCA (Sarco/Endoplasmic Reticulum Ca2+ -ATPase) pump. By restoring calcium homeostasis within the ER lumen, researchers observed a significant recalibration of pro-insulin folding, effectively "re-tooling" the cell for physiological rather than pathological secretion.
Furthermore, pharmacological scaffolding using Glucagon-like Peptide-1 Receptor Agonists (GLP-1 RAs) has emerged as a critical therapeutic pillar. Beyond their incretin effect, these agents engage the cAMP/PKA and PI3K/Akt signalling pathways, which promote beta-cell survival and stimulate the proliferation of existing mass. Crucially, recent studies indexed in *The Lancet* highlight that GLP-1 RAs induce "beta-cell rest" by improving insulin sensitivity and reducing the basal secretory demand. This period of secretory quiescence allows the intracellular machinery to clear misfolded protein aggregates through enhanced autophagy, a cellular "housekeeping" process that is often suppressed in the state of hyperinsulinaemic exhaustion.
At the frontier of INNERSTANDIN research is the concept of re-differentiation. Exhausted beta-cells often lose their mature identity, reverting to a progenitor-like state (dedifferentiation) characterised by the loss of FoxO1 activity and the expression of markers such as Neurogenin3. Recovery protocols must therefore aim to "re-identity" these cells. Current evidence suggests that aggressive glycaemic control—achieved through a combination of caloric restriction and SGLT2 inhibition—can create a metabolic environment conducive to the re-expression of MafA and PDX1, the transcription factors requisite for mature beta-cell function. By removing the glucotoxic insult, we facilitate the anatomical restoration of the islet’s cytoarchitecture, ensuring that the pancreas is not merely managed, but functionally resuscitated.
Summary: Key Takeaways
Pancreatic beta-cell exhaustion represents a terminal failure of the islet architecture to maintain homeostatic equilibrium against relentless metabolic insulance. At INNERSTANDIN, we expose the reality that this pathophysiology is not merely a quantitative loss of cell mass, but a qualitative collapse of cellular identity. Data from the UK Prospective Diabetes Study (UKPDS) indicates that functional capacity often diminishes by approximately 50% prior to clinical diagnosis, driven by the synergistic assault of glucotoxicity and lipotoxicity. This metabolic overreach precipitates chronic endoplasmic reticulum (ER) stress; the resulting proteostatic burden triggers the Unfolded Protein Response (UPR). When these compensatory mechanisms are overwhelmed, cells transition from adaptive hypertrophy to pro-apoptotic signalling via the CHOP pathway.
Furthermore, exhaustion is characterised by cellular dedifferentiation—a phenotypic regression where beta-cells lose critical transcription factor signatures (such as MafA and NeuroD1) and revert to a non-functional, progenitor-like state. This process is compounded by the cytotoxic accumulation of islet amyloid polypeptide (IAPP) and mitochondrial dysfunction secondary to chronic reactive oxygen species (ROS) generation. Ultimately, the anatomy of exhaustion reveals a systemic prioritisation of cellular survival over endocrine function, rendering traditional secretagogue therapies increasingly futile as the biological machinery for insulin biosynthesis fundamentally disintegrates. Peer-reviewed research, including longitudinal cohorts in *The Lancet*, confirms that once the FoxO1-mediated stress response is compromised, the path toward irreversible glycaemic instability becomes structurally encoded within the pancreatic niche.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Beta-cell exhaustion is characterized by a loss of mature identity and a shift toward a dedifferentiated state under metabolic pressure.
Dedifferentiation of pancreatic beta-cells is a primary mechanism for the decline in insulin production during the progression of type 2 diabetes.
Prolonged exposure to high glucose levels induces endoplasmic reticulum stress that significantly impairs the structural integrity of beta-cells.
Genetic and environmental factors converge on beta-cell regulatory networks to accelerate the loss of functional mass in metabolic disease.
Chronic hyperinsulinemia leads to mitochondrial exhaustion and oxidative stress, providing an anatomical basis for beta-cell failure.
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "The Anatomy of Pancreatic Beta-Cell Exhaustion"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Anatomy — products curated by our research team for educational relevance and biological support.

Magnesium L-Threonate
Magnesium Blend – The Most Important Mineral

Energy Blend Supports
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper