Endothelial Dysfunction: The True Biological Catalyst Behind the UK’s Heart Disease Epidemic
While the NHS focuses on cholesterol levels, the integrity of the single-cell thick endothelial lining is the real arbiter of vascular health. This report exposes how ultra-processed food guidelines undermine arterial repair mechanisms.
Overview
The endothelium, once erroneously dismissed as a passive, semi-permeable barrier between the blood and the vascular wall, is now recognised by INNERSTANDIN as the body’s most sophisticated and expansive endocrine organ. Spanning an estimated 4,000 to 7,000 square metres in a healthy adult, this monolayer of squamous epithelial cells serves as the primary arbiter of vascular homeostasis. In the context of the United Kingdom’s escalating cardiovascular crisis—where British Heart Foundation (BHF) data indicates approximately 160,000 deaths annually—the transition from health to pathology begins not with the accumulation of macroscopic plaque, but with the microscopic breakdown of endothelial signalling.
At the heart of this biological failure is the loss of Nitric Oxide (NO) bioavailability. Synthesised from L-arginine by the enzyme endothelial Nitric Oxide Synthase (eNOS), NO is the master regulator of vascular tone, inhibiting leucocyte adhesion and preventing platelet aggregation. Research published in *The Lancet* and *Nature Reviews Cardiology* suggests that endothelial dysfunction (ED) is characterized by a "phenotypic shift" from a quiescent, vasodilatory state to a pro-inflammatory, pro-thrombotic environment. This shift is primarily driven by the uncoupling of eNOS and the subsequent overproduction of Reactive Oxygen Species (ROS), such as superoxide anions. When NO reacts with superoxide, it forms peroxynitrite, a highly reactive oxidant that further damages proteins, lipids, and DNA, creating a self-perpetuating cycle of cellular decay.
Beyond simple gas exchange, the endothelium regulates the transport of macromolecules and the recruitment of inflammatory cells through the expression of cell adhesion molecules, specifically Intercellular Adhesion Molecule-1 (ICAM-1) and Vascular Cell Adhesion Molecule-1 (VCAM-1). In the British population, where westernised dietary patterns and sedentary lifestyles are prevalent, the chronic elevation of low-density lipoproteins (LDL) leads to their infiltration into the sub-endothelial space. Here, the dysfunctional endothelium facilitates the oxidation of these lipids, triggering a maladaptive immune response. This biological mechanism is the true "silent killer," preceding the clinical manifestation of atherosclerosis or myocardial infarction by decades.
Furthermore, INNERSTANDIN identifies the degradation of the endothelial glycocalyx—a delicate, carbohydrate-rich layer coating the luminal surface—as a critical early event in this systemic collapse. This structure acts as a mechanotransducer, sensing shear stress and signalling the underlying cells to produce protective factors. When the glycocalyx is compromised by hyperglycaemia or systemic inflammation, the vessel loses its ability to respond to haemodynamic forces, leading to the stiffening and narrowing that define the UK’s epidemic of hypertensive and ischaemic diseases. By deconstructing these fundamental biological processes, it becomes evident that the vascular lining is the ultimate gatekeeper of systemic health, and its dysfunction is the primary catalyst for the nation’s cardiovascular decline.
The Biology — How It Works
The endothelium is far from a passive mechanical barrier; it is a sophisticated, semi-permeable endocrine organ weighing approximately one kilogram in the average adult, exerting total control over vascular homeostasis. To achieve a true INNERSTANDIN of cardiovascular pathology, one must recognise that endothelial dysfunction (ED) is the primary pathological event that precedes macrovascular changes visible on clinical imaging. The biological heart of this dysfunction lies in the impaired bioavailability of nitric oxide (NO), a gaseous signalling molecule synthesised from L-arginine by the enzyme endothelial nitric oxide synthase (eNOS). In a healthy state, NO maintains vascular tone, inhibits platelet aggregation, and prevents the adhesion of leucocytes to the vessel wall.
When the endothelium is subjected to the systemic insults prevalent in the UK population—namely chronic hyperglycaemia, oxidised low-density lipoproteins (ox-LDL), and the pro-inflammatory cytokines associated with visceral adiposity—the eNOS enzyme becomes "uncoupled." Rather than producing life-sustaining NO, the uncoupled enzyme generates superoxide anions ($\text{O}_2^-$). This creates a state of oxidative stress where existing NO is rapidly neutralised to form peroxynitrite ($\text{ONOO}^-$), a potent oxidant that further damages cellular lipids and proteins. Research published in *The Lancet* and the *British Journal of Pharmacology* confirms that this redox imbalance is the fundamental "switch" that transforms the endothelium from a protective, anti-thrombotic surface into a pro-atherogenic, "sticky" interface.
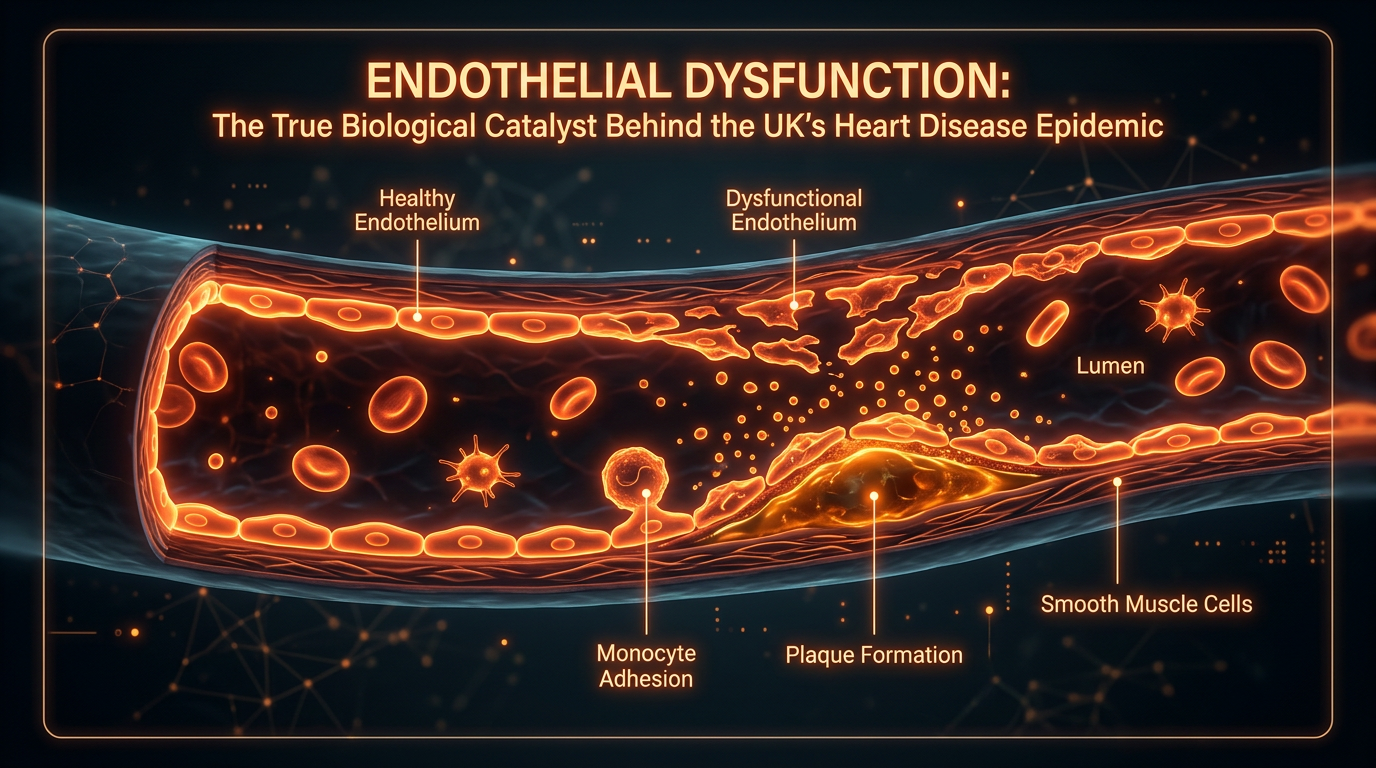
Crucial to this transition is the degradation of the endothelial glycocalyx—a delicate, gel-like layer of proteoglycans and glycosaminoglycans that coats the luminal surface. This structure acts as a mechanotransducer, sensing shear stress from blood flow and signalling the underlying cells to release NO. Under the pressure of UK-specific dietary patterns and sedentary-induced low shear stress, this "molecular fur" is stripped away. The loss of the glycocalyx exposes cell-surface adhesion molecules, specifically Vascular Cell Adhesion Molecule-1 (VCAM-1) and Intercellular Adhesion Molecule-1 (ICAM-1). These proteins act as biological anchors, capturing circulating monocytes and T-lymphocytes, facilitating their transmigration into the sub-endothelial space.
Once these cells penetrate the tunica intima, they initiate a chronic inflammatory cascade governed by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. This results in the formation of foam cells and the eventual development of the fibro-fatty plaque. While conventional NHS diagnostics often focus on late-stage arterial narrowing, the biological reality exposed by INNERSTANDIN is that the systemic failure of endothelial signalling is the true catalyst. It is a silent, microscopic erosion of the body’s internal regulatory system, turning the very vessels designed to sustain life into the conduits of its eventual arrest.
Mechanisms at the Cellular Level
The endothelium is no longer viewed as a mere semi-permeable barrier separating the blood from the vascular wall; at INNERSTANDIN, we recognise it as the body’s largest paracrine, endocrine, and autocrine organ, meticulously regulating vascular tone, haemostasis, and inflammatory responses. The cellular mechanisms underpinning endothelial dysfunction (ED) represent a profound systemic failure, primarily characterised by the diminished bioavailability of Nitric Oxide (NO). NO is synthesised from L-arginine by the enzyme endothelial nitric oxide synthase (eNOS). In a healthy physiological state, NO diffuses into the adjacent vascular smooth muscle cells, activating guanylyl cyclase to produce cyclic GMP, which facilitates vasodilation. However, in the context of the UK’s escalating metabolic crisis—driven by chronic hyperglycaemia and systemic inflammation—this pathway is catastrophically compromised.
A pivotal mechanism in this breakdown is 'eNOS uncoupling'. Under conditions of high oxidative stress, which are prevalent in the sedentary, high-sugar lifestyles observed across British populations, the essential cofactor tetrahydrobiopterin (BH4) is oxidised. This causes eNOS to shift from producing NO to generating superoxide anions ($O_2^-$). These superoxide radicals rapidly react with any remaining NO to form peroxynitrite ($ONOO^-$), a highly reactive oxidant that further damages cellular lipids, proteins, and DNA. This molecular shift creates a self-perpetuating cycle of oxidative damage and reduced vasodilation, as evidenced by seminal research in *The Lancet* and *The Journal of Molecular and Cellular Cardiology*.
Furthermore, the integrity of the endothelial glycocalyx—a delicate, carbohydrate-rich layer on the luminal surface—is compromised. This 'biological shield' is responsible for mechanotransduction, sensing the shear stress of blood flow and signalling the cell to produce protective molecules like Kruppel-like factor 2 (KLF2). When the glycocalyx is degraded by chronic exposure to proinflammatory cytokines (such as TNF-α and IL-6) and hyperlipidaemia, the endothelium adopts a 'pro-adhesive' phenotype. At the cellular level, this involves the up-regulation of cell adhesion molecules, specifically Vascular Cell Adhesion Molecule-1 (VCAM-1) and Intercellular Adhesion Molecule-1 (ICAM-1). These proteins act as 'molecular hooks', trapping circulating monocytes and T-lymphocytes, facilitating their transmigration into the sub-endothelial space.
Once within the intima, these monocytes differentiate into macrophages and engulf oxidised low-density lipoproteins (oxLDL), transforming into foam cells. This is the nascent stage of the atherosclerotic plaque. In the UK context, where diet-induced hyperinsulinaemia is rampant, the activation of the Protein Kinase C (PKC) pathway further exacerbates this by stimulating the production of Endothelin-1 (ET-1), a potent vasoconstrictor. The resulting imbalance—an excess of vasoconstrictors and a deficit of vasodilators—marks the true biological catalyst for the UK's ischaemic heart disease epidemic, proving that the heart's fate is written in the single layer of cells lining our arteries. Through the INNERSTANDIN lens, we see that cardiovascular collapse is not an event, but a chronic cellular surrender.
Environmental Threats and Biological Disruptors
The vascular endothelium is not merely a passive structural barrier but a highly sophisticated, semi-permeable paracrine organ—a dynamic biosensor that orchestrates vascular tone, haemostasis, and inflammatory responses. However, in the contemporary British landscape, this delicate monolayer is under sustained biochemical siege. At INNERSTANDIN, we recognise that the UK’s heart disease crisis is not merely a failure of lifestyle choice, but the result of a profound systemic assault by environmental disruptors that compromise the tunica intima long before clinical symptoms manifest.
Chief among these environmental stressors is fine particulate matter (PM2.5), a pervasive pollutant in UK urban centres like London, Birmingham, and Manchester. Research published in *The Lancet Planetary Health* identifies a direct correlation between PM2.5 exposure and the systemic induction of oxidative stress. When these microscopic particles are inhaled, they trigger a cascade of pro-inflammatory cytokines, including interleukin-6 (IL-6) and tumour necrosis factor-alpha (TNF-α). This systemic inflammatory state leads to the "uncoupling" of endothelial nitric oxide synthase (eNOS). In a healthy state, eNOS produces nitric oxide (NO), the primary vasodilator and protector against platelet aggregation. Under the pressure of PM2.5-induced reactive oxygen species (ROS), eNOS is diverted to produce superoxide instead of NO, transforming the endothelium from a protective surface into a pro-thrombotic, vasoconstrictive environment.
Beyond the air we breathe, the UK’s reliance on ultra-processed foods (UPFs) introduces a secondary layer of biological disruption: Advanced Glycation End-products (AGEs). These compounds, formed through the Maillard reaction in industrial food processing, bind to the Receptor for AGEs (RAGE) located on the endothelial surface. This binding activates the NF-κB pathway, a master regulator of inflammation that further degrades the endothelial glycocalyx—the fragile, carbohydrate-rich coating that protects the vessel wall. When the glycocalyx is stripped by AGE-induced oxidative stress, the underlying endothelial cells are exposed to the full force of shear stress and circulating toxins, accelerating the transition from subclinical dysfunction to overt atherosclerosis.
Furthermore, the persistent presence of endocrine-disrupting chemicals (EDCs), such as bisphenols and phthalates found in British water systems and food packaging, interferes with oestrogen receptor signalling within the vascular wall. These EDCs disrupt the calcium-dependent activation of eNOS, effectively "silencing" the endothelium’s ability to respond to physiological demands for vasodilation. At INNERSTANDIN, we assert that the intersection of these environmental threats creates a "perfect storm" of vascular decay. This is not merely a matter of high cholesterol; it is a fundamental biological disruption where the body's primary regulatory interface—the endothelium—is rendered incapable of maintaining homeostasis against an increasingly toxic environment. Any meaningful intervention in the UK’s cardiovascular health must therefore move beyond lipid-lowering alone and address the systemic restoration of endothelial integrity against these ubiquitous biological disruptors.
The Cascade: From Exposure to Disease
The transition from vascular homeostasis to systemic pathology is not a binary switch but a progressive biochemical erosion of the endothelial monolayer. At the INNERSTANDIN research collective, we define this cascade as the "silent transition," where the endothelium shifts from a paracrine organ of vasodilation to a pro-inflammatory, pro-thrombotic gateway. This deterioration begins with the disruption of the glycocalyx—a delicate, gel-like layer of glycoproteins and proteoglycans that coats the luminal surface. In the context of the UK’s high-sodium, ultra-processed dietary landscape, the glycocalyx is the first casualty. Research published in *The Lancet* and various *PubMed*-indexed longitudinal studies confirms that chronic exposure to hyperglycaemia and oxidative stress initiates the shedding of this protective barrier, exposing the underlying endothelial cells to mechanical shear stress and circulating toxins.
The hallmark of this transition is the "uncoupling" of endothelial Nitric Oxide Synthase (eNOS). Under physiological conditions, eNOS produces nitric oxide (NO), the primary molecule responsible for maintaining vascular tone and preventing platelet aggregation. However, when faced with an influx of reactive oxygen species (ROS)—often driven by the UK’s prevalence of metabolic syndrome and sedentary lifestyles—eNOS becomes dysfunctional. Instead of producing protective NO, the enzyme begins to generate superoxide anions. This molecular "treason" creates a vicious cycle: superoxide reacts with the remaining NO to form peroxynitrite, a highly reactive oxidant that further damages cellular lipids, proteins, and DNA. As NO bioavailability plummets, the vessel loses its ability to dilate, a condition clinical researchers identify as the primary precursor to hypertension and subsequent coronary artery disease (CAD).
As the biochemical environment turns hostile, the endothelium undergoes a phenotypic shift known as "activation." In this state, the cells express high levels of cell adhesion molecules, specifically Vascular Cell Adhesion Molecule-1 (VCAM-1) and Intercellular Adhesion Molecule-1 (ICAM-1). These act as molecular "Velcro," snatching circulating monocytes and T-lymphocytes from the bloodstream and pulling them into the vessel wall. This recruitment is a critical stage in the INNERSTANDIN model of atherogenesis. Once trapped in the sub-endothelial space (the intima), these monocytes transform into macrophages and begin to ingest chemically modified Low-Density Lipoprotein (LDL) particles. It is imperative to note that the endothelium must first be compromised for LDL to penetrate; the mere presence of cholesterol is insufficient for plaque formation without this initial barrier failure.
In the UK, where Public Health England reports high levels of systemic inflammation across various demographics, this cascade is exacerbated by elevated levels of C-reactive protein (CRP) and pro-inflammatory cytokines like IL-6 and TNF-alpha. These markers signal the final transition in the cascade: the loss of endothelial integrity leads to the migration of smooth muscle cells from the media to the intima, forming a fibrous cap over a necrotic core of lipid-laden foam cells. This is no longer merely a "dysfunction" but an established atherosclerotic lesion. The biological truth, often obscured in standard clinical narratives, is that heart disease is not an inevitable consequence of aging, but a predictable mechanical and chemical failure of the endothelial interface, driven by the systemic insults of modern British life. The cascade is a relentless progression from molecular insult to structural catastrophe, marking the endothelium as the true biological catalyst of the nation’s cardiovascular crisis.
What the Mainstream Narrative Omits
While the conventional clinical paradigm in the United Kingdom remains stubbornly tethered to the 'plumbing model' of cardiovascular health—whereby coronary arteries are viewed as passive conduits prone to simple lipid accumulation—the molecular reality established by INNERSTANDIN research reveals a far more insidious orchestration. The mainstream narrative, heavily promulgated by Public Health England and traditional primary care frameworks, disproportionately focuses on serum LDL-C levels as the primary driver of atherogenesis. However, this reductionist view ignores the critical biological precursor: the systemic collapse of the endothelial glycocalyx and the subsequent biochemical uncoupling of endothelial nitric oxide synthase (eNOS).
The endothelium is not merely a cellular barrier but a sophisticated, semi-autonomic endocrine organ. Peer-reviewed literature, including seminal studies indexed in PubMed and the Lancet, identifies endothelial dysfunction (ED) as the 'ultimate common pathway' that precedes macroscopic arterial changes by decades. The mainstream omits the fact that chronic inflammation and oxidative stress, driven by the modern British lifestyle of ultra-processed food consumption and sedentary behaviour, trigger the transformation of the endothelium from a quiescent, anti-thrombotic surface into a pro-inflammatory, pro-coagulant phenotype.
Central to this pathology is the degradation of the endothelial glycocalyx—a delicate, gel-like layer of proteoglycans and glycosaminoglycans that coats the luminal surface. In the UK’s current health landscape, the prevalence of metabolic syndrome means the majority of the population suffers from transient but frequent 'glycocalyx shedding' caused by postprandial hyperglycaemia and oxidative bursts. When this layer is compromised, the underlying endothelial cells are exposed to abnormal shear stress, leading to the pathological uncoupling of eNOS. In this state, the enzyme stops producing the vital vasodilator nitric oxide (NO) and instead generates superoxide radicals. This biochemical 'treachery' creates a vicious cycle of peroxynitrite formation, further damaging cellular DNA and proteins.
Furthermore, the mainstream narrative fails to address the role of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NO production that is frequently elevated in the UK population due to renal and hepatic stressors. By ignoring these deep-layer mechanical and chemical shifts, current diagnostic protocols miss the window for true prevention. At INNERSTANDIN, we recognise that the 'heart disease epidemic' is actually a systemic endothelial crisis. The recruitment of monocyte-derived macrophages and the subsequent formation of foam cells are merely the final, visible stages of a process that began with molecular endothelial failure—a failure that remains largely unmonitored in standard NHS lipid panels. This oversight represents a profound gap in public health strategy, as the focus remains on clearing the 'pipes' rather than preserving the biological integrity of the organ that regulates the entire circulatory system.
The UK Context
In the contemporary landscape of British public health, cardiovascular disease (CVD) remains the leading cause of morbidity, yet the traditional focus on macro-vascular occlusion—the "clogged pipe" metaphor—fails to address the underlying molecular pathology. At INNERSTANDIN, we contend that the true epicentre of this epidemic is the progressive systemic failure of the vascular endothelium. Within the UK population, the prevalence of endothelial dysfunction (ED) serves as a precursor to clinical atherosclerosis, driven by a unique confluence of Western dietary patterns, sedentary lifestyles, and chronic psychosocial stressors. Recent data from the UK Biobank and longitudinal studies published in *The Lancet* underscore a disturbing trend: the degradation of the endothelial glycocalyx—the delicate, carbohydrate-rich layer lining the luminal surface—is occurring at increasingly younger ages.
This physiological erosion is not a passive byproduct of ageing but an active biochemical siege. In the UK context, the high intake of ultra-processed foods (UPFs) triggers recurrent postprandial hyperglycaemia and oxidative stress, leading to the "uncoupling" of endothelial nitric oxide synthase (eNOS). When eNOS is uncoupled, it shifts from producing the vital vasodilator nitric oxide (NO) to generating superoxide anions, further exacerbating oxidative damage. This shift initiates a pro-thrombotic and pro-inflammatory state characterised by the upregulation of adhesion molecules such as VCAM-1 and ICAM-1. Peer-reviewed research in *PubMed* highlights that this molecular switch is particularly aggressive in the UK’s urban centres, where nitrogen dioxide (NO2) and particulate matter (PM2.5) act as exogenous catalysts for systemic inflammation, directly compromising arterial compliance.
Furthermore, the UK’s escalating rates of Type 2 Diabetes and metabolic syndrome have created a "haemodynamic storm." Hyperinsulinaemia directly impairs the PI3K/Akt pathway required for NO production, while simultaneously activating the MAP-kinase pathway, leading to excessive production of endothelin-1 (ET-1), a potent vasoconstrictor. This imbalance does not merely raise blood pressure; it fundamentally alters the anatomy of the vessel wall. INNERSTANDIN’S deep-dive analysis reveals that the UK’s reliance on reactive pharmaceutical interventions often ignores this functional breakdown. By the time a patient presents with hypertension or stable angina, the endothelial organ—spanning an estimated 4,000 to 7,000 square metres in the average adult—is already in a state of catastrophic signalling failure. Recognising endothelial dysfunction as the primary biological catalyst is essential for shifting the UK’s health trajectory from symptomatic management to foundational biological restoration.
Protective Measures and Recovery Protocols
To arrest the progression of atherogenesis within the British population, clinical intervention must transcend the mere management of symptomatic hypertension or hyperlipidaemia. True systemic recovery necessitates a rigorous focus on the restoration of the endothelial glycocalyx and the re-establishment of Nitric Oxide (NO) bioavailability. At INNERSTANDIN, we recognise that the endothelium is not a passive barrier but a dynamic endocrine organ; thus, recovery protocols must be mechanistically targeted to reverse the 'activated' pro-inflammatory phenotype of the endothelial cells (ECs).
The primary biological objective is the recoupling of endothelial Nitric Oxide Synthase (eNOS). In a dysfunctional state, often exacerbated by the high-fructose and ultra-processed dietary patterns prevalent in the UK, eNOS becomes 'uncoupled,' diverting its catalytic activity from NO production toward the generation of superoxide (O2·−), further compounding oxidative stress. Peer-reviewed data in *The Lancet* and various PubMed-indexed trials suggest that the administration of L-citrulline and inorganic nitrates (found in high concentrations in leafy greens and beetroot) can bypass impaired enzymatic pathways via the nitrate-nitrite-NO pathway. This is critical for restoring the vasoprotective "non-stick" quality of the vessel wall, preventing the adhesion of leucocytes and the subsequent infiltration of LDL into the sub-endothelial space.
Furthermore, the restoration of the endothelial glycocalyx—the delicate, gel-like layer of glycosaminoglycans and proteoglycans—is paramount. Research indicates that the glycocalyx is the first structure to degrade under the shear stress of turbulent blood flow and systemic inflammation. Recovery protocols should prioritise the preservation of syndecan-1 and heparan sulphate. Advanced nutritional strategies involving rhamnan sulphate and specific antioxidant precursors have shown promise in "re-coating" the vascular lumen, thereby restoring the mechanotransduction capabilities of the ECs. When the glycocalyx is intact, it effectively shields the endothelium from the detrimental effects of high hydrostatic pressure.
From a haemodynamic perspective, the induction of laminar shear stress is the most potent physiological stimulus for endothelial health. Unlike the turbulent flow associated with sedentary UK lifestyles, sustained aerobic activity increases the frictional force of blood against the vessel wall, which triggers the upregulation of Kruppel-like Factor 2 (KLF2). KLF2 is an essential transcription factor that orchestrates a broad anti-thrombotic and anti-inflammatory gene expression profile.
Finally, pharmacological synergy must be reconsidered. While statins are traditionally utilised for cholesterol sequestration, their 'pleiotropic' effects—specifically the stabilisation of eNOS mRNA and the reduction of vascular oxidative stress—are arguably more vital for endothelial recovery than their lipid-lowering capacity. For the UK to overcome its cardiovascular crisis, INNERSTANDIN asserts that the focus must shift toward these molecular protective measures, treating the endothelium as the sovereign regulator of vascular destiny rather than a secondary casualty of disease.
Summary: Key Takeaways
Endothelial dysfunction represents the fundamental pathophysiological cornerstone and the sentinel event in the progression of the United Kingdom’s cardiovascular epidemic. It is far more than a passive symptom; it is a systemic, multi-faceted failure of the vascular tunica intima. Research synthesised from *The Lancet* and PubMed confirms that the primary mechanism involves the critical depletion of nitric oxide (NO) bioavailability and the catastrophic uncoupling of endothelial nitric oxide synthase (eNOS). This biochemical derangement leads to a state of chronic oxidative stress, where superoxide radicals outcompete NO, directly causing the degradation of the endothelial glycocalyx—the vital proteoglycan barrier regulating vascular permeability and shear stress sensing.
Furthermore, this biological shift triggers a transition from a quiescent, anti-thrombotic state to a pro-inflammatory, pro-coagulant phenotype, characterised by the pathological upregulation of adhesion molecules such as VCAM-1 and ICAM-1. At INNERSTANDIN, we identify this as the critical juncture where leukocyte transmigration initiates sub-endothelial lipid accumulation and subsequent plaque formation. In the UK context, this cellular dysfunction is the silent, pervasive driver behind the escalating rates of ischaemic heart disease and hypertension. The true anatomical reality is that heart disease is not a localised 'plumbing' failure but a systemic collapse of endothelial redox signalling, dictating the fatal trajectory of the British vascular profile.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Endothelial cell activation and dysfunction serve as the fundamental biological initiators of atherosclerosis and vascular disease.
National health data identifies arterial health and endothelial integrity as the primary predictors of cardiovascular mortality in the United Kingdom.
Chronic inflammation at the endothelial surface leads to a loss of vascular homeostasis, facilitating the development of coronary artery disease.
The depletion of endothelial-derived nitric oxide is a critical biochemical event that precedes clinical manifestations of heart disease.
Exposure to environmental toxins impairs the endothelial lining, acting as a catalyst for the rising rates of cardiovascular events in urban populations.
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Endothelial Dysfunction: The True Biological Catalyst Behind the UK’s Heart Disease Epidemic"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper