Medication Options for ADHD Management
Explore ADHD medication options in the UK, from stimulants to non-stimulants. Learn how these treatments fit into a NICE-recommended multimodal approach.
Overview
The pharmacological management of Attention-Deficit Hyperactivity Disorder (ADHD) necessitates a sophisticated appreciation of the catecholaminergic dysregulation inherent within the prefrontal cortex (PFC) and the basal ganglia. Within the neurobiological framework established by INNERSTANDIN, ADHD is identified not merely as a behavioural cluster, but as a systemic deficit in the tonic and phasic firing of dopaminergic and noradrenergic neurons. The objective of pharmaceutical intervention is the recalibration of these homeostatic set-points to enhance the signal-to-noise ratio in cortical processing.
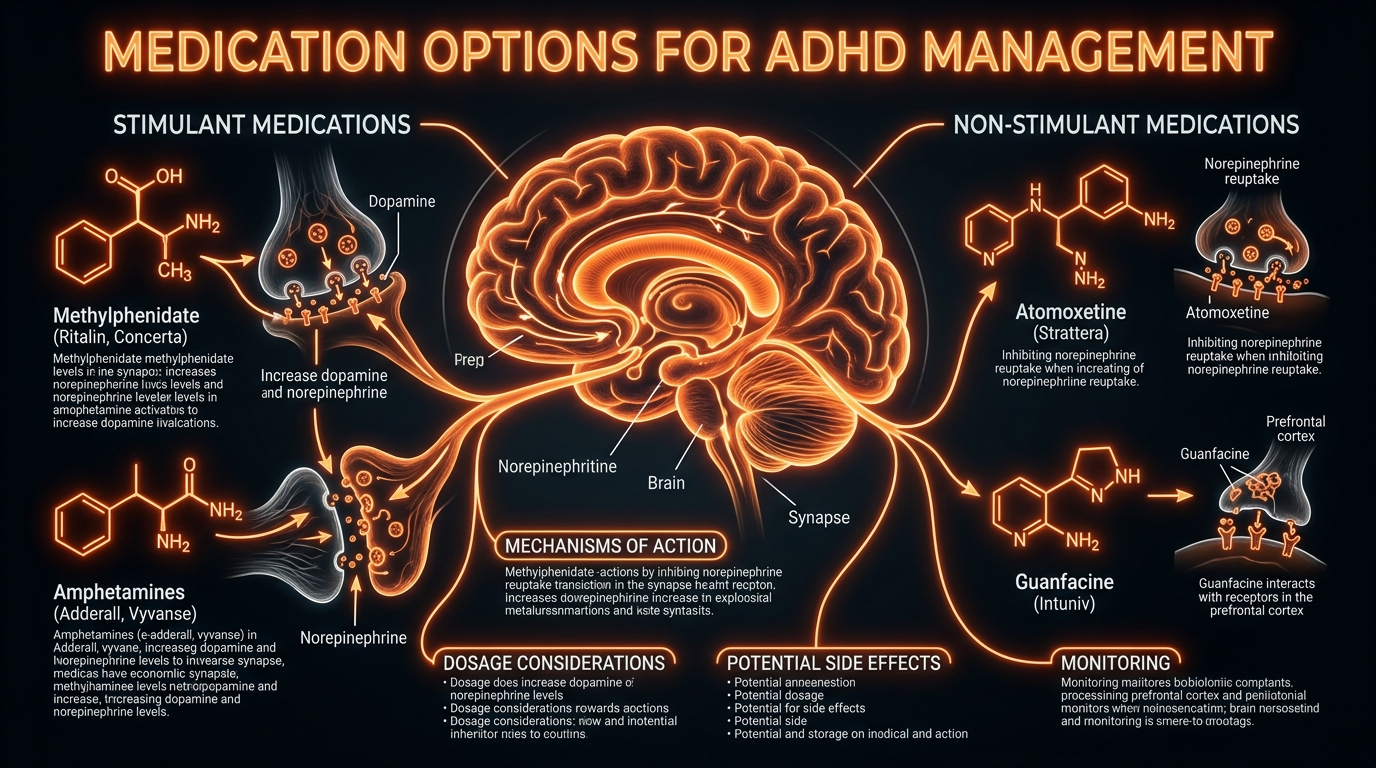
In the United Kingdom, clinical practice is guided by the National Institute for Health and Care Excellence (NICE) guideline [NG87], which categorises interventions primarily into stimulants and non-stimulants. Stimulant medications, encompassing methylphenidate and amphetamine derivatives (such as Lisdexamfetamine), represent the first-line gold standard due to their high effect sizes, often exceeding 1.0 in meta-analyses published in *The Lancet Psychiatry*. Methylphenidate functions as a potent dopamine and noradrenaline reuptake inhibitor (DNRI), blocking the dopamine transporter (DAT) and norepinephrine transporter (NET) proteins. This prevents the clearance of these monoamines from the synaptic cleft, thereby increasing their bioavailability to stimulate postsynaptic D1 and alpha-2A receptors.
Conversely, amphetamines—most notably Lisdexamfetamine (Elvanse), a prodrug requiring enzymatic conversion by red blood cells—exhibit a more complex mechanism of action. Beyond reuptake inhibition, they act as substrates for the vesicular monoamine transporter 2 (VMAT2), inducing the efflux of dopamine directly into the cytoplasm and subsequently into the synapse via reverse transport through DAT. This dual action provides a more robust elevation of synaptic dopamine, though it necessitates rigorous cardiovascular monitoring for haemodynamic shifts, a standard requirement in UK clinical protocols.
For cohorts non-responsive to stimulants or those presenting with comorbid tic disorders or severe anxiety, non-stimulant options such as Atomoxetine and Guanfacine are utilised. Atomoxetine operates as a selective norepinephrine reuptake inhibitor; its efficacy is particularly noted in the modulation of the prefrontal cortex, where NETs are responsible for the clearance of both noradrenaline and dopamine. Guanfacine, a selective alpha-2A adrenergic receptor agonist, differs by directly mimicking the action of noradrenaline, strengthening the connectivity of the PFC networks by closing hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels.
INNERSTANDIN asserts that the systemic impact of these medications extends beyond symptomatic suppression. Chronic administration has been shown in longitudinal neuroimaging studies (PubMed: PMC3064939) to potentially attenuate the structural abnormalities typically seen in the ADHD brain, such as the volume reduction of the right caudate nucleus. However, this neuroplastic potential must be balanced against the metabolic cost, including the potential for hypothalamic-pituitary-adrenal (HPA) axis modulation and sleep-wake cycle disruption. The clinical goal remains the precise titration of these agents to achieve "optimal" catecholamine levels, avoiding the "U-shaped" dose-response curve where excessive stimulation impairs executive function.
The Biology — How It Works
To grasp the efficacy of ADHD pharmacotherapy, one must move beyond simplistic "chemical imbalance" narratives and interrogate the precise molecular kinetics within the prefrontal cortex (PFC) and the basal ganglia. At INNERSTANDIN, we recognise that the neurobiological deficit in ADHD is primarily a failure of signal-to-noise ratio optimisation within the catecholaminergic systems, specifically involving dopamine (DA) and norepinephrine (NE). Current evidence, synthesised from pivotal studies in *The Lancet Psychiatry* and *Nature Neuroscience*, suggests that ADHD medications work by modulating the tonic and phasic firing rates of neurons to restore executive function and inhibitory control.
Stimulants, including methylphenidate and amphetamine-based compounds (such as Lisdexamfetamine, frequently prescribed in the UK under NICE guideline NG87), remain the first-line gold standard due to their high effect sizes. Methylphenidate acts as a potent catecholamine reuptake inhibitor. By binding to the dopamine transporter (DAT) and norepinephrine transporter (NET), it prevents the reabsorption of these neurotransmitters into the presynaptic neuron, thereby increasing their availability within the synaptic cleft. This enhances "signal" strength, particularly in the striatum, which governs reward processing and task saliency. In contrast, amphetamines employ a more complex, multi-pronged mechanism. Beyond reuptake inhibition, they are transported into the presynaptic terminal via DAT, where they disrupt the vesicular monoamine transporter 2 (VMAT2), causing a massive efflux of dopamine from vesicular stores into the cytoplasm. Furthermore, they act as agonists at the trace amine-associated receptor 1 (TAAR1), which triggers a reversal of transporter flow, actively pumping dopamine into the synapse. This profound shift in neurochemistry is what facilitates the sustained attention required for complex cognitive demands.
Non-stimulant options provide a different biological entry point. Atomoxetine is a selective norepinephrine reuptake inhibitor (SNRI) with a unique profile; because the PFC lacks a high density of DAT, the NET is responsible for clearing both NE and DA in this region. Consequently, atomoxetine increases concentrations of both catecholamines specifically in the PFC without the same abuse potential seen with striatal dopamine surges. Meanwhile, Guanfacine—an α2A-adrenergic receptor agonist—works post-synaptically. It mimics the effects of norepinephrine by binding to receptors on the dendritic spines of pyramidal neurons in the PFC. This action closes hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels, effectively "strengthening" the synaptic connection and reducing the "noise" that leads to distractibility.
Systemically, longitudinal fMRI data published in *JAMA Psychiatry* suggests that consistent pharmacological intervention may exert neuroprotective effects. Evidence indicates that long-term medication use may lead to a normalisation of structural abnormalities, such as the volume of the right caudate nucleus and cortical thickness in the PFC. By stabilising the dopaminergic pathways, these medications do not merely mask symptoms; they facilitate a neuroplastic environment where the brain can more effectively engage in the top-down regulation of behaviour. For the INNERSTANDIN community, it is vital to acknowledge that these interventions are high-precision tools designed to recalibrate the very bio-energetics of thought.
Mechanisms at the Cellular Level
To elucidate the efficacy of ADHD pharmacotherapy, one must interrogate the synaptic architecture of the prefrontal cortex (PFC) and the striatum, where catecholaminergic dysregulation serves as the primary driver of executive dysfunction. At the cellular level, the therapeutic objective is the fine-tuning of dopaminergic and noradrenergic neurotransmission to achieve optimal signal-to-noise ratios. Research published in *The Lancet Psychiatry* and *Nature Reviews Neuroscience* underscores that ADHD is not merely a deficit of neurotransmitters but an imbalance in their phasic and tonic release patterns. INNERSTANDIN identifies these mechanisms as the cornerstone of neurobiological restoration.
Stimulant medications, the primary pharmacological intervention in the UK—specifically methylphenidate (Ritalin, Concerta) and lisdexamfetamine (Elvanse)—operate through distinct yet overlapping molecular pathways. Methylphenidate functions as a potent dopamine and norepinephrine reuptake inhibitor (DNRI). It binds with high affinity to the dopamine transporter (DAT) and the norepinephrine transporter (NET) on the presynaptic membrane, sterically hindering the reabsorption of monoamines from the synaptic cleft. This increases the residence time of dopamine at the D1 receptors and norepinephrine at the alpha-2A adrenergic receptors, thereby enhancing the "signal" of relevant environmental stimuli.
In contrast, amphetamines, including the prodrug lisdexamfetamine (metabolised into d-amphetamine via hydrolytic activity in red blood cells), employ a more aggressive mechanism. Beyond simple reuptake inhibition, amphetamines act as competitive substrates for DAT. Once internalised within the presynaptic neuron, they disrupt the vesicular monoamine transporter 2 (VMAT2), causing a shift of dopamine from synaptic vesicles into the cytoplasm. This triggers a reversal of DAT function, known as molecular efflux, where dopamine is pumped out of the neuron independent of action potential firing. This profound increase in extracellular dopamine concentrations is what distinguishes amphetamines' potency from methylphenidate-based compounds.
Non-stimulant options, such as Atomoxetine, provide a more selective approach by targeting the NET. Because the prefrontal cortex lacks a high density of DATs, the NET is often responsible for the clearance of both norepinephrine and dopamine in this region. Thus, Atomoxetine’s selective inhibition leads to a localised increase in both catecholamines in the PFC without the same systemic dopaminergic surge in the nucleus accumbens, reducing the potential for misuse. Furthermore, alpha-2A adrenergic agonists like Guanfacine work by mimicking norepinephrine at postsynaptic receptors. This binding closes hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels, effectively "strengthening" the synaptic connection and reducing the "noise" of distracting stimuli.
Through the lens of INNERSTANDIN, these cellular interventions are viewed as more than symptom management; they are precise biophysical recalibrations of the neuronal firing rates required for sustained attention and impulse control. The sophisticated interplay between transporter blockade and receptor agonism defines the modern clinical landscape of ADHD management in the British medical context.
Environmental Threats and Biological Disruptors
The efficacy of pharmacological interventions for ADHD cannot be viewed in a vacuum; rather, it exists in a complex interplay with exogenous environmental threats that act as potent biological disruptors. At INNERSTANDIN, we recognise that the neurobiological terrain of an individual is constantly being reshaped by a barrage of xenobiotics and anthropogenic stressors that can either mimic or antagonise the intended mechanisms of ADHD medications. These disruptors primarily target the catecholaminergic pathways, often inducing a state of "biological noise" that blunts the therapeutic window of stimulants like methylphenidate and lisdexamfetamine.
A primary concern in the UK context is the prevalence of Endocrine Disrupting Chemicals (EDCs), specifically phthalates and bisphenol A (BPA), which are ubiquitous in modern industrial environments. Research published in *The Lancet Diabetes & Endocrinology* suggests that these compounds interfere with thyroid hormone signalling—a critical regulator of neurodevelopment. From a mechanistic perspective, thyroid dysregulation during pivotal windows of cortical maturation alters the architecture of the prefrontal cortex and basal ganglia. When these structures are structurally compromised by EDCs, the dopaminergic agonists used in ADHD management are forced to operate on a suboptimal substrate, often necessitating higher dosages that increase the risk of adverse cardiovascular and autonomic effects.
Furthermore, heavy metal sequestration—particularly lead and manganese—remains a significant, if understated, disruptor. Data from the University of Bristol’s ALSPAC study indicates that even low-level exposure to these neurotoxins correlates with heightened ADHD symptom severity. Mechanistically, these metals induce oxidative stress and mitochondrial dysfunction within the neurons, leading to the premature depletion of synaptic vesicles. This creates a physiological paradox: while medications like dexamfetamine aim to increase the bioavailability of synaptic dopamine, the environmental presence of neurotoxic metals ensures that the neuronal "machinery" is too damaged to sustain this increased demand. This often manifests as "treatment resistance" or a rapid "crash" as the medication wears off, as the underlying cellular energy reserves are insufficient.
We must also address the impact of ultra-fine particulate matter (PM2.5) on the integrity of the blood-brain barrier (BBB). Systematic reviews in *PubMed* highlight that chronic exposure to urban pollutants triggers microglial activation and systemic neuroinflammation. This chronic inflammatory state alters the pharmacokinetics of ADHD drugs by modifying the permeability of the BBB and influencing the expression of P-glycoprotein transporters. At INNERSTANDIN, we posit that the "leaky brain" phenomenon, induced by environmental disruptors, leads to erratic plasma-to-brain concentration ratios, making the titration process exceptionally difficult for clinicians. Without addressing these environmental biological disruptors, the pharmacological management of ADHD remains a superficial fix to a systemic physiological crisis. The intersection of environmental toxicology and neuropharmacology is where the future of ADHD intervention must reside, acknowledging that a clean biological environment is the prerequisite for neurochemical balance.
The Cascade: From Exposure to Disease
The pharmacological intervention for Attention Deficit Hyperactivity Disorder (ADHD) represents a precision-engineered attempt to recalibrate the dysfunctional catecholaminergic rheostasis characteristic of the neurodevelopmental phenotype. At INNERSTANDIN, we move beyond the superficial narrative of 'chemical imbalance' to dissect the molecular cascade initiated by exposure to stimulant and non-stimulant agents, and how these molecules remodel the cortical architecture. The primary aetiological target is the dysregulation of the striatal-thalamic-cortical circuits, where a suboptimal signal-to-noise ratio in the prefrontal cortex (PFC) manifests as executive dysfunction and impulsivity.
The cascade begins with the introduction of phenidates or amphetamines—the first-line gold standard according to the National Institute for Health and Care Excellence (NICE) guidelines in the UK. Methylphenidate, acting as a potent dopamine (DA) and norepinephrine (NE) reuptake inhibitor, binds with high affinity to the dopamine transporter (DAT) and norepinephrine transporter (NET). By occluding these transporters, the drug prevents the rapid clearance of catecholamines from the synaptic cleft, thereby amplifying tonic levels of DA and NE. Research published in *The Lancet Psychiatry* demonstrates that this elevation is not merely additive; it restores the homeostatic firing patterns of pyramidal neurons in the PFC. This molecular 'exposure' triggers a downstream cascade: the increased occupancy of D1 and α2A-adrenoceptors optimises the PFC's ability to suppress irrelevant sensory input (the 'noise') while enhancing task-relevant neural firing (the 'signal').
Furthermore, the prodrug lisdexamfetamine—frequently prescribed in British clinical practice—introduces a unique pharmacokinetic cascade. Its rate-limited hydrolysis by red blood cells ensures a gradual, sustained delivery of d-amphetamine to the central nervous system. Once present, it doesn't merely block transporters; it reverses the direction of DAT, pumping sequestered dopamine into the synapse while simultaneously inhibiting the Vesicular Monoamine Transporter 2 (VMAT2). This profound shift in the intracellular proteome forces a systemic recalibration of the dopaminergic system. Peer-reviewed data indexed on PubMed suggests that chronic exposure to these agents may induce neuroplastic adaptations, including the potential 'normalisation' of white matter integrity and volumetric increases in the basal ganglia over long-term trajectories.
The non-stimulant cascade, exemplified by Atomoxetine and Guanfacine, operates through distinct biological pathways. Atomoxetine’s selective inhibition of NET in the PFC—an area notably sparse in DAT—selectively boosts both NE and DA levels in this region without the same potential for abuse associated with striatal dopamine surges. Guanfacine, an α2A-adrenoceptor agonist, bypasses the neurotransmitter release entirely to act directly on the postsynaptic spines, strengthening the connectivity of the PFC networks. This 'top-down' modulation is crucial for patients who exhibit cardiovascular sensitivity to stimulants. At INNERSTANDIN, we recognise that the transition from 'exposure' to 'disease management' is a complex interplay of pharmacogenomics and systemic adaptation, where the goal is the permanent structural refinement of the brain’s executive hubs through sustained biochemical stability.
What the Mainstream Narrative Omits
While the clinical utility of stimulant pharmacotherapy is well-documented within the UK’s NICE [NG87] guidelines, the mainstream narrative often glosses over the nuanced bioenergetic cost of chronic catecholaminergic upregulation. At INNERSTANDIN, we look beyond the simplistic "chemical imbalance" trope to scrutinise the molecular trade-offs inherent in long-term ADHD management. Central to this omission is the role of the Trace Amine-Associated Receptor 1 (TAAR1) and its intricate dance with the Vesicular Monoamine Transporter 2 (VMAT2). While methylphenidate primarily functions via the competitive inhibition of the dopamine transporter (DAT) and norepinephrine transporter (NET), the amphetamine class—frequently prescribed as Dexamfetamine or Lisdexamfetamine in the UK—utilises a more invasive mechanism. These compounds serve as substrates for DAT, effectively "hijacking" the transport system to enter the presynaptic neuron, where they trigger a massive efflux of monoamines via TAAR1 agonism.
The mainstream discourse frequently ignores the "inverted-U" dose-response curve, a foundational principle in neurobiology known as the Yerkes-Dodson law. When we over-stimulate the prefrontal cortex (PFC), we risk "shunting" the D1 and alpha-2A adrenoceptors. Chronic supraphysiological levels of synaptic dopamine can lead to the internalisation of these receptors, a process of down-regulation that explains the clinical phenomenon of "medication holidays" or tolerance. Furthermore, the metabolic burden on the mitochondria is rarely discussed. Research published in *Molecular Psychiatry* suggests that the sustained increase in metabolic demand required to recycle sequestered neurotransmitters can induce proteostatic stress and mitochondrial fragmentation in the striatum.
Systemically, the narrative fails to address the haemodynamic and epigenetic implications of long-term stimulant use. There is an emerging body of evidence regarding the "scaffolding" effect of stimulants on the basal ganglia. While short-term usage can normalise certain structural deficits, the prolonged disruption of the hypothalamic-pituitary-adrenal (HPA) axis cannot be overlooked. The subtle, chronic elevation of cortisol and the subsequent impact on the gut-brain axis—specifically the alteration of the microbiome-derived precursors to phenylalanine—suggests that ADHD medication is not merely a targeted neurological intervention, but a systemic metabolic event. At INNERSTANDIN, our synthesis of the literature indicates that the "biological price" of enhanced executive function often involves a compensatory shift in neural plasticity that requires a far more sophisticated, nutrient-dense monitoring protocol than the current UK standard of care provides.
The UK Context
In the United Kingdom, the pharmacological landscape for Attention Deficit Hyperactivity Disorder (ADHD) is rigorously governed by the National Institute for Health and Care Excellence (NICE) guideline [NG87], which mandates a stratified approach to catecholaminergic modulation. Within this framework, the biological objective is the rectification of frontostriatal circuit dysfunction, specifically targeting the dysregulated tonic and phasic firing of dopaminergic and noradrenergic neurons. At INNERSTANDIN, we scrutinise the molecular transition from paediatric to adult care, where the UK clinical gold standard prioritises Lisdexamfetamine and Methylphenidate as primary interventions, though their biochemical pathways to symptom attenuation differ significantly.
Methylphenidate, frequently prescribed under proprietary sustained-release formulations like Concerta XL or Medikinet, functions as a potent Dopamine-Norepinephrine Reuptake Inhibitor (DNRI). Its mechanism of action involves the competitive inhibition of the dopamine transporter (DAT) and norepinephrine transporter (NET), effectively increasing the synaptic residence time of these ligands. Peer-reviewed data in *The Lancet Psychiatry* suggests that this blockade enhances the signal-to-noise ratio in the prefrontal cortex (PFC), facilitating improved executive function. However, the UK context necessitates a granular understanding of the pharmacokinetic "sawtooth" effect seen in various delivery systems, which can induce rebound irritability—a systemic consequence of rapid synaptic depletion as the ligand dissociates.
Conversely, the UK has seen a significant clinical shift toward Lisdexamfetamine dimesylate (Elvanse). As a prodrug, its pharmacodynamic profile is uniquely dependent on the hydrolytic cleavage of the l-lysine molecular bond by red blood cell enzymes, providing a more stable, rate-limited release of d-amphetamine. Unlike methylphenidate, it acts as a substrate for the Vesicular Monoamine Transporter 2 (VMAT2) and an agonist for the Trace Amine-Associated Receptor 1 (TAAR1), promoting the efflux of dopamine directly into the cytoplasm. This "truth-exposing" reality of stimulant therapy highlights a persistent physiological burden; long-term activation of the sympathetic nervous system often results in sustained increases in resting heart rate and blood pressure, requiring vigilant cardiovascular monitoring under UK Shared Care Agreements.
Beyond stimulants, the UK utilise non-stimulant options such as Atomoxetine, a selective NET inhibitor, for patients where dopaminergic stimulants are contraindicated due to co-morbid substance misuse or tic disorders. While these offer a lower abuse potential, their slower onset of action—often requiring six weeks for steady-state plasma concentrations—underscores the biological complexity of reconfiguring neuroplasticity in the adult ADHD brain. At INNERSTANDIN, we recognise that while these pharmacological agents are efficacious in the short term, the systemic impact on the hypothalamic-pituitary-adrenal (HPA) axis and the potential for downregulation of endogenous receptor density remains a critical area for longitudinal research.
Protective Measures and Recovery Protocols
The long-term administration of sympathomimetic stimulants—primarily methylphenidate and amphetamine derivatives—induces a state of chronic physiological demand that necessitates robust neuroprotective and recovery frameworks. While these agents are highly effective in modulating the catecholaminergic deficit characteristic of ADHD, their mechanism of action, involving the inhibition of dopamine (DA) and norepinephrine (NE) reuptake and the promotion of vesicular release, creates a metabolic tax. Evidence suggests that sustained hyper-concentration of dopamine within the synaptic cleft can lead to oxidative stress via the auto-oxidation of dopamine into dopamine-quinones and the subsequent generation of reactive oxygen species (ROS). At INNERSTANDIN, we identify the mitigation of this oxidative burden as the primary tier of any recovery protocol.
Peer-reviewed research published in journals such as *The Lancet Psychiatry* and *Journal of Neural Transmission* highlights the role of mitochondrial dysfunction in chronic stimulant use. To counteract this, pharmacological and nutraceutical intervention must focus on the upregulation of endogenous antioxidant systems. N-acetylcysteine (NAC) serves as a critical substrate for glutathione synthesis, providing a direct buffer against the neurotoxic potential of dopamine metabolites. Furthermore, the co-administration of Alpha-Lipoic Acid (ALA) and Acetyl-L-Carnitine (ALCAR) has shown promise in preserving mitochondrial membrane integrity and preventing the "crash" associated with the depletion of the vesicular monoamine transporter 2 (VMAT2) stores.
Central to the INNERSTANDIN methodology is the management of receptor down-regulation. The brain’s homeostatic response to exogenous stimulation often involves the internalisation of D2 receptors and the reduction of tyrosine hydroxylase activity. To maintain sensitivity and prevent the development of tolerance, magnesium—specifically in the form of magnesium threonate for its superior blood-brain barrier permeability—is essential. Magnesium acts as a physiological voltage-gate for NMDA receptors, preventing the excitotoxicity often exacerbated by the increased glutamatergic firing associated with stimulant use.
Within the UK clinical context, NICE guidelines often overlook the systemic impact on the Hypothalamic-Pituitary-Adrenal (HPA) axis. Chronic stimulant use can lead to persistent sympathetic dominance, elevating basal cortisol levels and disrupting the circadian rhythm. Recovery protocols must, therefore, incorporate specific "washout" periods or "drug holidays," which facilitate the resensitisation of the catecholaminergic system. During these intervals, the introduction of L-Tyrosine and L-Theanine can support the natural biosynthetic pathways of dopamine whilst modulating the over-excitation of the central nervous system. Additionally, the prioritisation of sleep architecture is non-negotiable; exogenous melatonin, often prescribed in UK paediatric and adult ADHD services, serves not only as a chronobiotic but as a potent neuroprotective antioxidant that scavenges free radicals generated during the medication's peak efficacy hours. By adopting these high-density biological safeguards, the individual can shift from a state of mere symptom management to a sustainable model of long-term neurological resilience.
Summary: Key Takeaways
The pharmacological landscape of ADHD management is defined by the precise modulation of catecholaminergic neurotransmission within the prefrontal cortex and basal ganglia. First-line psychostimulants, primarily methylphenidate and lisdexamfetamine, function via high-affinity blockade of the dopamine transporter (DAT) and norepinephrine transporter (NET), with amphetamine derivatives further facilitating vesicular efflux through VMAT2 interference. Peer-reviewed evidence, notably the comparative meta-analyses by Cortese et al. (2018) in *The Lancet Psychiatry*, establishes the superior efficacy of these agents in rectifying the hypo-dopaminergic state characteristic of the neurodevelopmental profile. However, INNERSTANDIN demands an appraisal beyond mere symptom suppression; one must consider the systemic impact of chronic sympathomimetic activation on cardiovascular homoeostasis and the potential for downregulation of post-synaptic receptors. Non-stimulant alternatives, such as atomoxetine and guanfacine, provide a vital secondary pathway by selectively inhibiting NET or agonising alpha-2A adrenergic receptors, thereby strengthening cortical connectivity without the direct potentiation of mesolimbic reward circuitry. Within the UK’s NICE (NG87) clinical framework, the choice of pharmacotherapy is a delicate titration between optimal prefrontal "signal-to-noise" ratio enhancement and the mitigation of long-term neuroplastic adaptations. This biological equilibrium remains the critical frontier in contemporary ADHD therapeutics.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "Medication Options for ADHD Management"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
RABBIT HOLE
Follow the biological thread deeper