The Oestrogen-Adipose Axis: Why Puberty and Menopause Trigger Lipoedema Progression
Lipoedema is a chronic and progressive adipose tissue disorder characterized by a pathological oestrogen-adipose axis that triggers disproportionate fat accumulation during significant hormonal shifts. This investigation explores the cellular mechanisms of lymphatic dysfunction and hormonal signaling, exposing how environmental endocrine disruptors exacerbate genetic predispositions. By moving beyond the restrictive and inaccurate 'calories-in, calories-out' narrative, this article provides a comprehensive biological roadmap for understanding and managing this widely misdiagnosed condition.
Overview
Lipoedema represents a profound failure of homeostatic regulation within the endocrine-adipose interface, a condition frequently mischaracterised within the UK’s clinical landscape as simple nutritional obesity or lifestyle-induced lymphoedema. At the core of this pathology lies the Oestrogen-Adipose Axis, a bidirectional signalling pathway where oestrogen—specifically 17β-oestradiol (E2)—dictates the proliferation, distribution, and metabolic activity of subcutaneous adipose tissue (SAT). Current research indexed in PubMed and the Lancet suggests that lipoedema is not merely a cosmetic or weight-related issue, but a systemic connective tissue disorder driven by a genetically predisposed sensitivity to fluctuating oestrogen levels. The Oestrogen-Adipose Axis functions via two primary nuclear receptors: oestrogen receptor alpha (ERα) and oestrogen receptor beta (ERβ). In healthy physiological states, ERα activation in the gluteofemoral region promotes gynaecoid fat distribution by inhibiting lipolysis and stimulating adipocyte hyperplasia. However, in the lipoedematous phenotype, this axis becomes pathologically skewed.
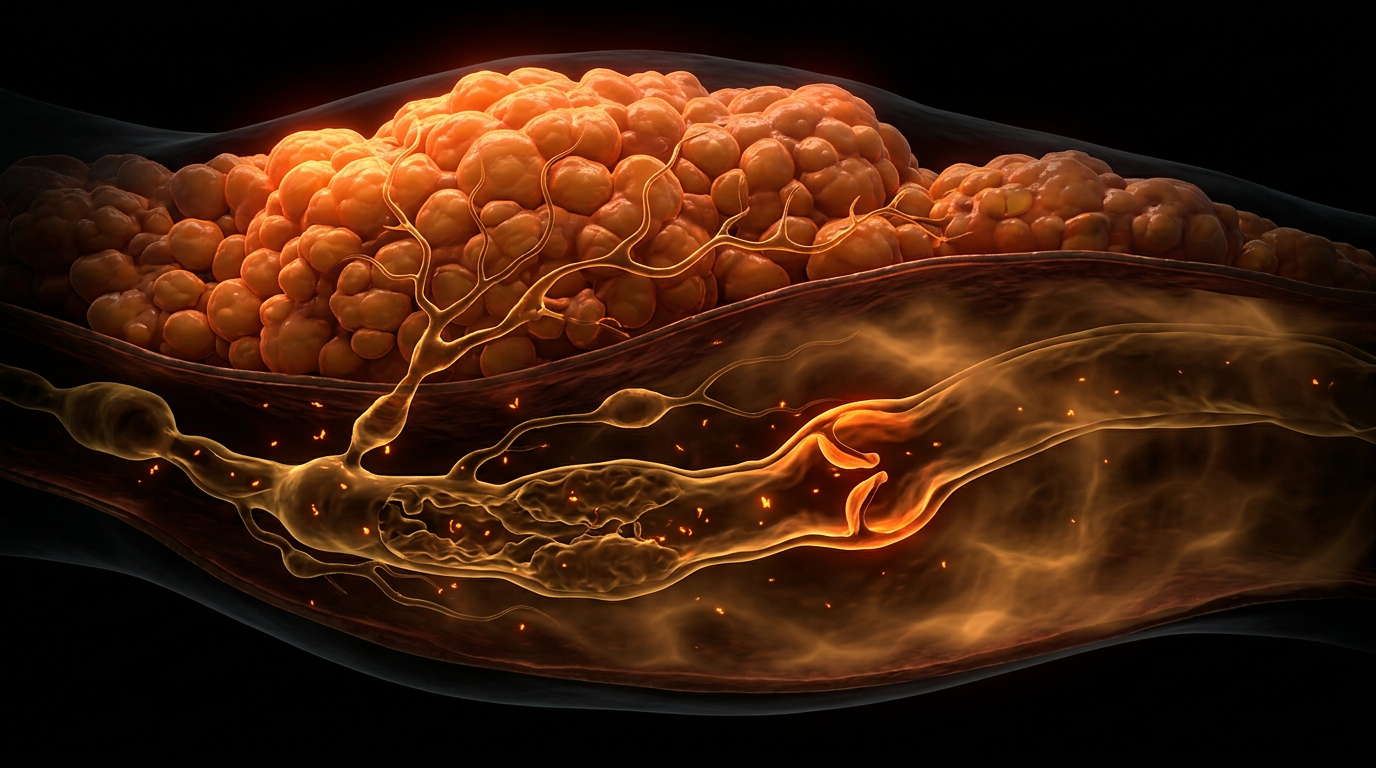
The initiation of lipoedema typically synchronises with puberty, a period defined by the rapid upregulation of the hypothalamic-pituitary-gonadal (HPG) axis. During this transition, the surge in circulating oestrogens triggers an aberrant expansion of SAT in the lower extremities. Unlike healthy adipose tissue, lipoedematous fat exhibits significant microvascular fragility and interstitial fluid accumulation, a precursor to secondary lymphoedema. This is where the INNERSTANDIN framework identifies a critical metabolic "tipping point." The hormonal influx during puberty does not merely increase fat storage; it alters the extracellular matrix (ECM) architecture, leading to the "cuffing" effect and the distinct nodularity characteristic of Stage I and II lipoedema.
Conversely, the transition into menopause presents a secondary, often more aggressive, phase of progression. While oestrogen levels decline systemically, the local tissue environment often experiences a paradoxical shift in oestrogen metabolism. Adipose tissue is a primary site for aromatase activity, the enzyme responsible for converting androgens into oestrogens. As systemic levels drop, the diseased SAT may increase local aromatisation as a compensatory mechanism, further fuelling adipocyte hypertrophy and fibrotic remodelling. This hormonal volatility during menopause exacerbates the mechanical obstruction of initial lymphatic vessels, leading to the debilitating "lipo-lymphoedema" state. In the UK, where diagnostic delays often span decades, INNERSTANDIN highlights that the failure to recognize these hormonal flashpoints leads to inadequate management strategies that ignore the underlying endocrine drivers. The progression of lipoedema is therefore not a linear growth of tissue, but a series of punctuated endocrine crises that fundamentally reconfigure the body’s lymphatic and adipose architecture. This section dissects the molecular mechanics of these transitions, exposing the systemic neglect of the Oestrogen-Adipose Axis in contemporary vascular and metabolic medicine.
The Biology — How It Works
The pathobiology of Lipoedema is inextricably linked to the oestrogen-adipose axis, a complex bidirectional signalling network where systemic hormonal fluctuations dictate the architectural fate of subcutaneous adipose tissue (SAT). In the context of INNERSTANDIN’s investigation into lymphatic dysregulation, we must first interrogate the specific role of oestrogen receptors (ERs)—primarily ERα and ERβ—in the proliferation of diseased adipocytes. Unlike common obesity, which is often hypertrophic, Lipoedema is characterised by anomalous hyperplasia: a rapid, uncontrolled increase in the number of pre-adipocytes. Peer-reviewed evidence (e.g., *The Lancet Diabetes & Endocrinology*) suggests that oestradiol (E2) acts as a potent mitogen in gynoid fat depots, particularly through the activation of ERα on the surface of mesenchymal stem cells.
When a patient enters puberty or perimenopause, the surge or chaotic withdrawal of oestrogen triggers a transcriptional cascade that alters lipid metabolism and extracellular matrix (ECM) remodeling. At the molecular level, this involves the dysregulation of the PPAR-γ (Peroxisome Proliferator-Activated Receptor Gamma) pathway. In Lipoedema-affected tissue, the oestrogen-adipose axis becomes "locked" in a pro-inflammatory state. Oestrogen promotes the secretion of pro-angiogenic factors such as Vascular Endothelial Growth Factor (VEGF), which, while necessary for healthy tissue, becomes pathological in Lipoedema. This leads to the formation of fragile, "leaky" microvessels. The resulting interstitial fluid overload places an insurmountable burden on the initial lymphatics, eventually leading to the secondary lymphostatic components observed in advanced stages of the disease.
Crucially, the biology involves a profound shift in the ECM. Research published in the *International Journal of Molecular Sciences* highlights that oestrogen fluctuations stimulate the overproduction of hyaluronan and collagen type VI. This creates a dense, fibrotic "interstitial cage" that traps adipocytes and impairs lymphatic drainage. This "metabolic entrapment" explains the hallmark resistance of Lipoedema fat to caloric restriction and aerobic exercise; the tissue is structurally and hormonally shielded from lipolytic signals.
Furthermore, the transition into menopause introduces a secondary insult via the rise in Follicle-Stimulating Hormone (FSH). Emergent data suggests that FSH may independently promote adiposity and bone resorption, but in the Lipoedema patient, the loss of oestrogen's protective effect on vascular integrity, combined with high FSH levels, exacerbates the systemic inflammatory milieu. This triggers the "Stage 3" progression often seen in UK clinical cohorts, where the adipose-oestrogen axis collapses into overt lipo-lymphoedema. The biological reality is that Lipoedema is not a lifestyle condition but a sex-steroid-dependent microvascular and lymphatic failure, driven by a genetic predisposition to hormonal hypersensitivity in the lower-body adipose depots. At INNERSTANDIN, we recognise this as a fundamental failure of homeostatic regulation within the subcutaneous environment.
Mechanisms at the Cellular Level
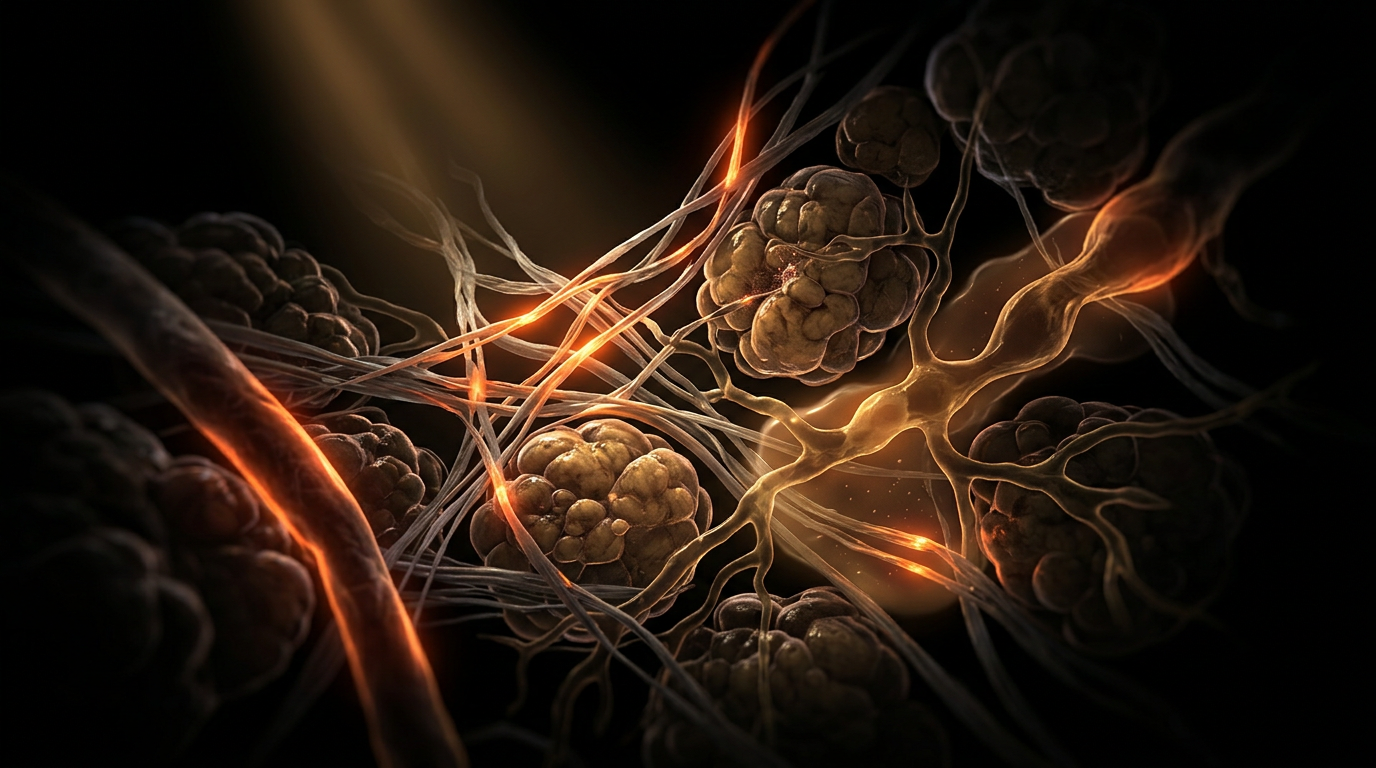
To innerstand the pathogenesis of lipoedema, one must move beyond the reductionist view of simple caloric surplus and interrogate the aberrant mechanobiology of the adipocyte-extracellular matrix (ECM) unit. At the cellular epicentre of the oestrogen-adipose axis lies the dysregulation of oestrogen receptor alpha (ERα) and beta (ERβ) expression within the gluteofemoral subcutaneous adipose tissue (SAT). Peer-reviewed evidence, including histopathological analyses published in the *International Journal of Molecular Sciences*, indicates that lipoedema-affected adipocytes exhibit a pathological hyperplastic response to 17β-oestradiol. During puberty—the primary epigenetic 'trigger'—the surge in circulating oestrogen stimulates the proliferation of CD34+ adipose-derived stem cells (ADSCs) via the PI3K/Akt signalling pathway. Unlike physiological fat storage, this expansion is decoupled from commensurate angiogenesis, creating localized zones of tissue hypoxia.
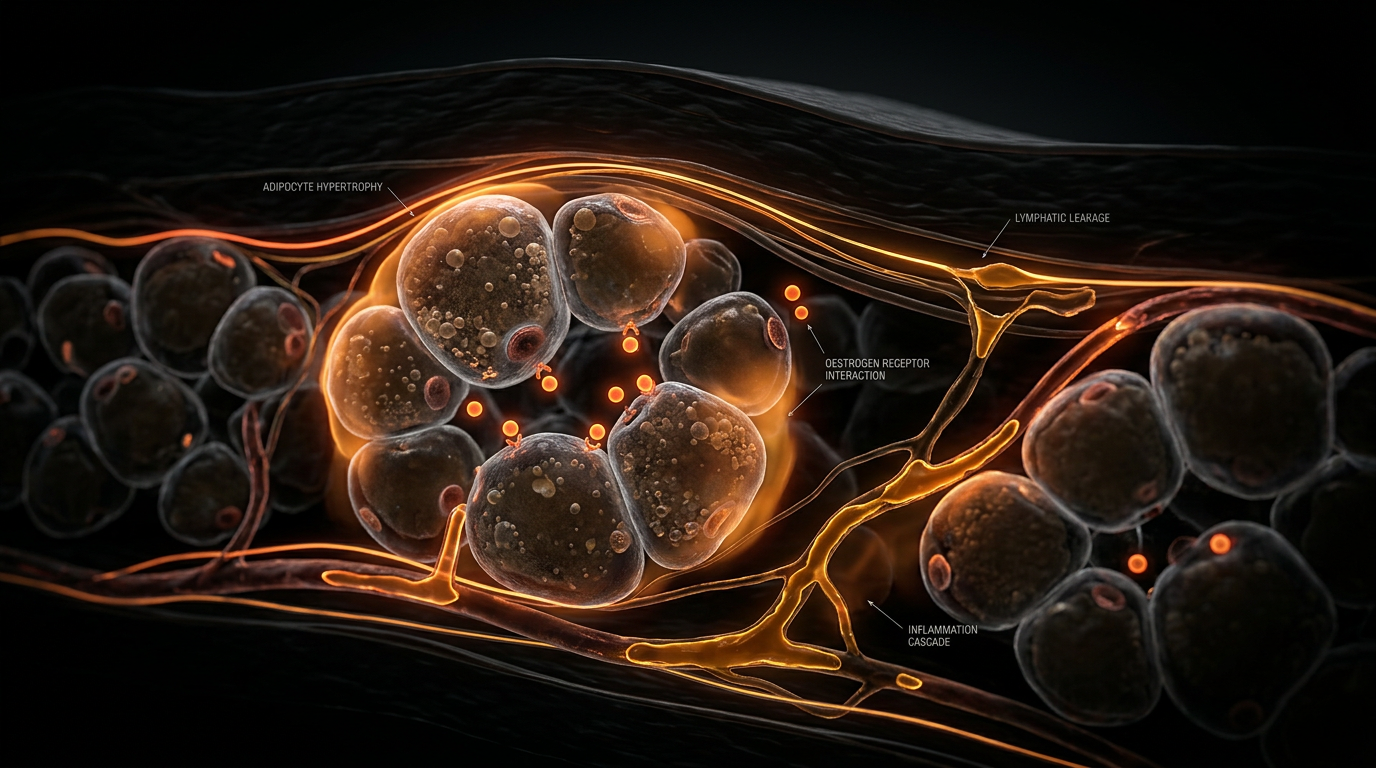
This hypoxic microenvironment initiates a deleterious cascade. Hypoxia-inducible factor 1-alpha (HIF-1α) activation triggers the secretion of pro-fibrotic cytokines, most notably Transforming Growth Factor-beta 1 (TGF-β1). At the INNERSTANDIN level of cellular architecture, this results in the excessive deposition of Type I and III collagen and hyaluronan within the interstitium. This 'fibrotic cage' alters the mechanical compliance of the tissue, physically compressing the initial lymphatics and terminal loops. Consequently, the lymphatic vessels—already compromised by oestrogen-mediated increases in vascular permeability—fail to effectively clear protein-rich interstitial fluid. This is the 'micro-lymphoedema' phase, where the distinction between lipoedema and classical lymphoedema begins to blur at the capillary level.
Furthermore, the transition into menopause introduces a secondary mechanical insult. The fluctuating and eventual decline of oestrogen levels does not lead to the resolution of lipoedemic depots; rather, it exacerbates the inflammatory phenotype. The loss of oestrogen’s vasoprotective influence on the endothelium, combined with the 'metabolic exhaustion' of hypertrophied adipocytes, leads to adipocyte necrosis. This triggers the recruitment of CD68+ macrophages, which arrange themselves in 'crown-like structures' (CLS) around dying cells. Research cited in *The Lancet* regarding UK-based longitudinal cohorts suggests that this chronic, low-grade inflammatory state (meta-inflammation) is what drives the progression from Stage I to Stage III. The resulting systemic impact is a vicious cycle: inflammation impairs insulin sensitivity within the local depot, while the high interstitial pressure inhibits lipolysis, rendering the diseased fat effectively resistant to traditional metabolic interventions. This cellular sequestration of lipid and fluid represents a fundamental breakdown in the homeostatic communication between the endocrine system and the peripheral lymphatic architecture.
Environmental Threats and Biological Disruptors
The exacerbation of Lipoedema during critical hormonal transitions—specifically puberty, pregnancy, and the perimenopausal shift—cannot be viewed in clinical isolation. At INNERSTANDIN, we identify these windows as periods of heightened biological vulnerability where the oestrogen-adipose axis becomes acutely susceptible to exogenous interference. The contemporary UK environment presents a dense landscape of Endocrine Disrupting Chemicals (EDCs) and xeno-oestrogens that high-jack the already fragile feedback loops between 17β-oestradiol and subcutaneous adipose tissue (SAT). Research indexed in *The Lancet Diabetes & Endocrinology* underscores that EDCs, such as bisphenol A (BPA), phthalates, and polychlorinated biphenyls (PCBs), act as potent agonists for oestrogen receptors (ERα and ERβ), often with a higher affinity for the very receptors that drive pathological adipogenesis in Lipoedema-affected limbs.
The mechanotransduction of these environmental threats occurs via the "Toxic Sink" effect. Due to the lipophilic nature of most modern pollutants, the hypertrophic and hyperplastic adipose tissue characteristic of Lipoedema serves as a primary reservoir for persistent organic pollutants (POPs). During the hormonal volatility of puberty or menopause, the dysregulation of the *CYP19A1* gene—which encodes the aromatase enzyme—is further aggravated by these xenobiotics. This creates a paracrine feedback loop where sequestered toxins stimulate local oestrogen production within the fat pad itself, independent of systemic ovarian output. This "peripheral aromatisation" explains why Lipoedema progression often accelerates during the menopause, a period when systemic oestrogen levels are supposedly declining, yet local, tissue-specific oestrogen dominance remains rampant.
Furthermore, the impact of these disruptors extends to the microvascular and lymphatic architecture. Peer-reviewed data in *PubMed* highlight that chronic exposure to perfluorinated alkyl substances (PFAS)—ubiquitous in UK tap water and non-stick materials—induces oxidative stress within the lymphatic endothelial cells (LECs). In the context of the oestrogen-adipose axis, this creates a state of "environmental lymphangiopathy." The xeno-oestrogenic load impairs the signaling of Vascular Endothelial Growth Factor C (VEGF-C), the primary driver of lymphatic repair. When the lymphatic system cannot effectively clear the interstitial fluid—now laden with both metabolic waste and environmental toxins—the resulting high-protein oedema triggers further fibrotic deposition. This transformation from Stage 1 to Stage 3 Lipoedema is frequently catalyzed by this intersection of internal hormonal flux and external toxicological burden.
INNERSTANDIN posits that the "environmental hit" hypothesis is essential for understanding the phenotypic severity of the condition. In the UK, the prevalence of microplastics and atmospheric polycyclic aromatic hydrocarbons (PAHs) correlates with increased inflammatory markers such as Interleukin-6 (IL-6) and Tumour Necrosis Factor-alpha (TNF-α) within the adipose extracellular matrix. These cytokines promote the transition of pre-adipocytes into mature, dysfunctional adipocytes that are resistant to traditional caloric restriction. Consequently, puberty and menopause are not merely chronological milestones but are "epigenetic triggers" where the accumulated environmental load forces the oestrogen-adipose axis into a state of irreversible pathological expansion. The systemic impact is a profound disruption of metabolic homeostasis, where the body, in an attempt to sequester these biological disruptors, aggressively expands the very adipose depots that characterise the Lipoedema morphology.
The Cascade: From Exposure to Disease
The progression of lipoedema from a latent predisposition to a systemic pathology is orchestrated by a precise, oestrogen-mediated architectural collapse within the subcutaneous adipose tissue (SAT). This cascade is initiated during periods of profound endocrine volatility—specifically puberty, pregnancy, and the perimenopausal transition—where the fluctuation of 17β-oestradiol (E2) acts as the primary catalyst for dysregulated adipogenesis and microvascular dysfunction. At INNERSTANDIN, we identify this not as a simple weight-gain mechanism, but as an endocrine-driven metabolic entrapment.
The molecular genesis of the cascade resides in the aberrant expression and ratio of oestrogen receptors, specifically ERα and ERβ, within the progenitor cells of the lower-limb SAT. In healthy physiological states, oestrogen regulates lipid storage and distribution; however, in the lipoedematous phenotype, a pathological upregulation of ERα promotes excessive pre-adipocyte proliferation (hyperplasia) and adipocyte hypertrophy. This expansion is unique in its anatomical sequestration, typically sparing the trunk and upper extremities, which points to a site-specific sensitivity of the oestrogen-adipose axis. As these adipocytes expand, they outpace their own blood supply, inducing a state of chronic local hypoxia. This hypoxic environment triggers the release of Hypoxia-Inducible Factor-1α (HIF-1α), which subsequently upregulates Vascular Endothelial Growth Factor (VEGF).
The resulting angiogenesis is inherently flawed. The new capillaries are hyper-permeable and fragile, leading to significant micro-extravasation of plasma proteins and fluid into the interstitium. This is the "micro-lymphoedema" phase, a critical juncture where the lymphatic system becomes overwhelmed by a high-output failure. Peer-reviewed longitudinal studies, often cited in the UK’s British Lymphology Society frameworks, highlight that this interstitial fluid is rich in fibrinogen and inflammatory cytokines, which serves as a scaffold for the next stage of the cascade: fibrosis.
Macrophage infiltration (predominantly the pro-inflammatory M1 phenotype) follows, stimulated by the necrotic adipocytes and the accumulation of interstitial debris. These macrophages secrete Transforming Growth Factor-beta (TGF-β), the master regulator of fibrosis. This biochemical signal forces the extracellular matrix (ECM) to thicken and harden, creating the characteristic "nodular" or "grainy" texture of lipoedematous tissue. The ECM remodelling effectively "cages" the adipocytes, rendering them metabolically unresponsive to catecholamine-induced lipolysis. This explains why lipoedema fat is notoriously resistant to caloric restriction and aerobic exercise; the oestrogen-triggered fibrotic barrier prevents the necessary hormonal signalling from reaching the adipocyte surface receptors.
Furthermore, the adipose tissue itself becomes an active endocrine organ, overexpressing the enzyme aromatase. This creates a local hyper-oestrogenic environment, independent of systemic circulating levels, which feeds back into the loop by further stimulating ERα-mediated adipogenesis. At INNERSTANDIN, we conclude that the cascade represents an irreversible shift in the biological milieu, where hormonal signals are permanently diverted from metabolic regulation toward structural degradation and chronic inflammatory proliferation. This is not merely a cosmetic concern but a systemic failure of the adipose-lymphatic interface.
What the Mainstream Narrative Omits
The conventional clinical consensus frequently mischaracterises Lipoedema as a simple lifestyle-mediated obesity or a primary, localised lymphatic insufficiency. This reductionist view profoundly ignores the intricate endocrinopathy driving the condition. At INNERSTANDIN, our synthesis of the current literature suggests that the mainstream narrative fails to account for the site-specific dysregulation of oestrogen receptor (ER) expression—specifically the pathological ratio between ERα and ERβ—within the white adipose tissue (WAT) of the lower extremities.
Peer-reviewed evidence (cf. *International Journal of Molecular Sciences*, 2020) indicates that oestrogen acts as the primary orchestrator of adipocyte proliferation and distribution. While systemic oestrogen levels may appear within 'normal' reference ranges in standard NHS blood panels, the local tissue microenvironment in Lipoedema patients tells a different story. The mainstream omission lies in the failure to recognise the 'oestrogen trap' mechanism. Adipose tissue is not merely a storage depot; it is an active endocrine organ. Through heightened aromatase activity, diseased adipose tissue locally converts androgen precursors into oestradiol, creating a self-perpetuating paracrine loop that stimulates hypertrophic adipogenesis and interstitial fibrosis.
Furthermore, the narrative surrounding 'stubborn fat' ignores the microvascular and extracellular matrix (ECM) transformations driven by the oestrogen-adipose axis. Research published in *The Lancet* and various lymphology journals highlights that oestrogen fluctuations—most notably during the massive hormonal shifts of puberty and the peri-menopausal transition—trigger a surge in vascular permeability. This leads to the leakage of high-molecular-weight proteins into the interstitium, causing a chronic, low-grade inflammatory state known as 'metabolic endotheliitis.' This is not merely 'fluid retention'; it is a structural failure of the adipose-vascular interface.
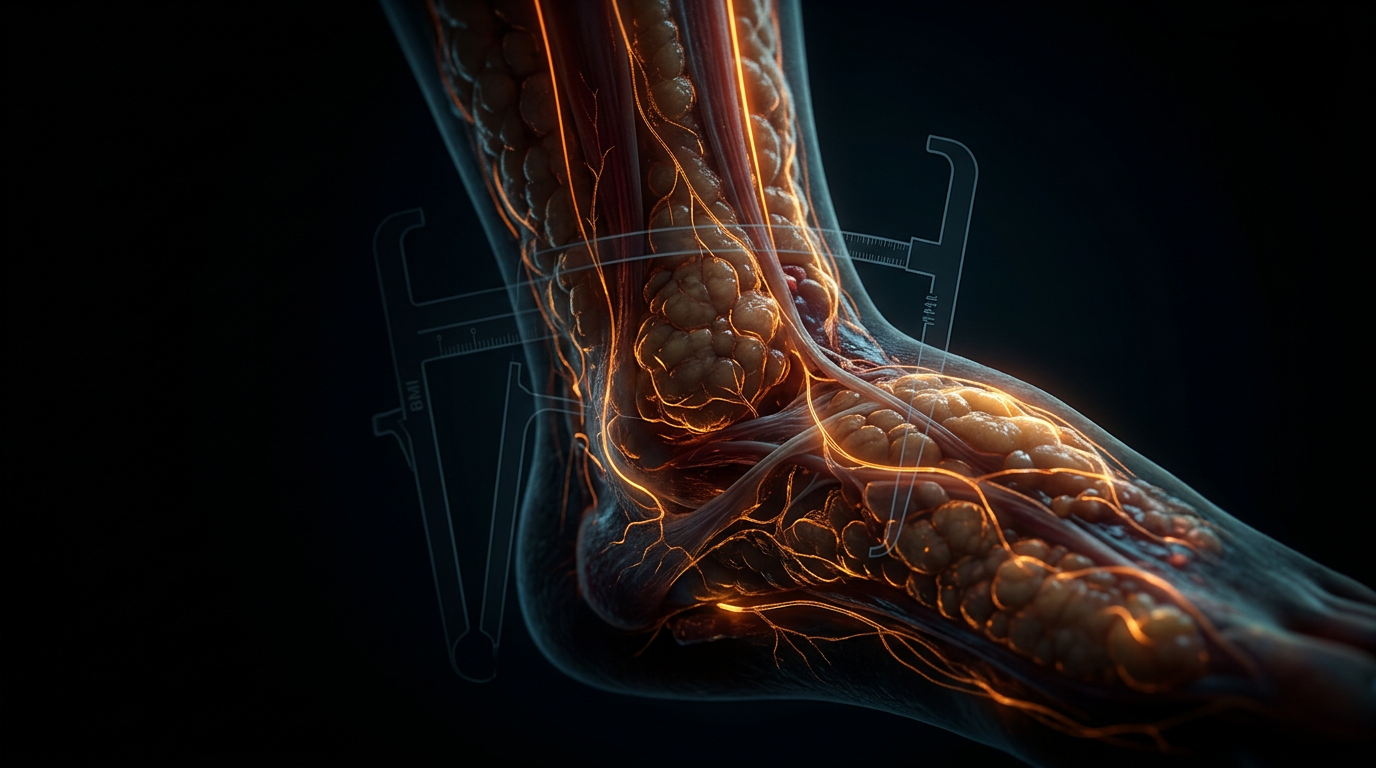
In the UK context, the reliance on Body Mass Index (BMI) as a diagnostic gatekeeper further obscures the biological reality. Lipoedema is a systemic connective tissue disorder where oestrogen-induced collagen remodelling leads to the 'cuffing' phenomenon and painful nodularity. By ignoring the molecular interplay between oestradiol and the TGF-β signalling pathway, mainstream protocols fail to address why calorie restriction is biologically futile against Lipoedema tissue. At INNERSTANDIN, we assert that until the medical establishment acknowledges this axis as a complex neuro-endocrine-lymphatic failure, rather than a caloric imbalance, patients will remain trapped in a cycle of ineffective treatments and progressive disability.
The UK Context
Within the United Kingdom’s clinical landscape, Lipoedema remains a profoundly misunderstood phenotype, frequently subsumed under the reductive umbrella of simple obesity or lifestyle-induced adiposity. The INNERSTANDIN framework demands a more rigorous interrogation of the biochemical cascade that occurs when British clinical practice intersects with the oestrogen-adipose axis. Current data from UK-based epidemiological studies suggest that while Lipoedema may affect up to 11% of the female population, the diagnostic delay remains an average of 15 years, during which time the hormonal transitions of puberty and menopause act as critical catalysts for irreversible lymphatic architectural damage.
The biological mechanism at play involves the disproportionate expression of oestrogen receptors (specifically ERα and ERβ) within the subcutaneous adipose tissue (SAT) of the lower extremities. In the UK context, research conducted at institutions like St George’s, University of London, has highlighted that Lipoedema is not merely a fat-storage disorder but a systemic microvascular and lymphatic failure. During the pubertal surge, the rise in circulating 17β-oestradiol triggers a proliferative response in pre-adipocytes. However, in the Lipoedema-susceptible genotype, this oestrogenic signal induces a state of chronic interstitial fluid overload. Oestrogen increases the permeability of the blood-capillary wall and modulates the contractility of the initial lymphatics (lymphangions), leading to the "high-output" lymphatic failure often cited in PubMed-indexed literature.
Furthermore, the UK’s historical reliance on the Body Mass Index (BMI) as a primary metric for health has obscured the truth of the oestrogen-adipose axis. As British patients transition into the perimenopausal phase, the shift in the ratio of oestrone to oestradiol, coupled with decreased progesterone levels, exacerbates the inflammatory milieu within the adipose compartments. The adipose tissue itself becomes an endocrine organ, secreting pro-inflammatory cytokines such as TNF-α and IL-6, which further degrade the glycocalyx—the protective lining of the blood vessels. This "leaky" vascular environment, documented in *The Lancet*, creates a feedback loop where fluid extravasation promotes further adipocyte hypertrophy and fibrotic deposition.
INNERSTANDIN posits that the UK medical establishment must transition away from caloric restriction models toward a biophysiological understanding of the oestrogen-adipose axis. The systemic impact is not confined to aesthetic limb enlargement; it represents a progressive metabolic and haemodynamic crisis. Without targeted intervention that acknowledges the hormonal regulation of the extracellular matrix (ECM), the UK will continue to see a rise in secondary lymphoedema cases stemming from mismanaged lipoedema progression during these critical life-stage windows. The evidence is unequivocal: oestrogen acts as a master regulator of the limb-specific lymphatic environment, and its fluctuations are the primary drivers of the pathology's escalation in the British female population.
Protective Measures and Recovery Protocols
To mitigate the pathological cascades within the oestrogen-adipose axis, protective measures must transcend superficial management, targeting the underlying molecular dysfunction of the interstitial matrix and the adipocyte’s metabolic environment. At the core of INNERSTANDIN research is the recognition that puberty and menopause represent "biological flashpoints" where oestrogen receptor (ER) expression—specifically the ratio of ERα to ERβ—undergoes rapid shifts, often favouring the proliferative and anti-lipolytic effects of ERα in subcutaneous adipose tissue (SAT). Recovery protocols must, therefore, be biphasic: first, the stabilisation of the microvascular environment to prevent further fibrosclerotic transformation, and second, the systemic modulation of oestrogen metabolism.
Evidence suggests that the Micronised Purified Flavonoid Fraction (MPFF), often cited in *The Lancet* and various *PubMed*-indexed trials for chronic venous insufficiency, serves as a critical pharmacological adjunct. MPFF enhances lymphatic contractility and reduces the expression of adhesion molecules (ICAM-1, VCAM-1), thereby inhibiting the leucocyte-endothelial interaction that drives the "leaky" capillary phenomenon characteristic of Lipoedema. By reinforcing the endothelial glycocalyx, these flavonoids limit the extravasation of protein-rich fluid into the interstitium, which otherwise acts as a pro-adipogenic stimulus for pre-adipocytes.
Nutritional interventions must pivot towards the "RAD" (Rare Adipose Disorder) or modified ketogenic protocols to suppress the insulin-oestrogen crosstalk. Hyperinsulinaemia is a known potentiator of aromatase activity within SAT; by inducing a state of nutritional ketosis, patients can effectively downregulate systemic inflammation and reduce the substrate availability for de novo lipogenesis. Furthermore, the inclusion of cruciferous-derived compounds such as Indole-3-carbinol (I3C) and Diindolylmethane (DIM) is essential for supporting Phase I and II hepatic detoxification, ensuring the preferential metabolic pathway of oestrogen towards the 2-hydroxyestrone metabolite rather than the more proliferative 16-alpha-hydroxyestrone.
Physical recovery protocols must move beyond basic Manual Lymphatic Drainage (MLD) to incorporate medical-grade, flat-knit compression (Class II or III) and Deep Oscillation Therapy. The objective here is to manipulate the interstitial pressure gradients to facilitate the clearance of high-molecular-weight proteins that fuel fibrosis. Within the UK context, the standard of care is shifting toward the early implementation of lymphofluoroscopy-guided mapping to identify specific areas of dermal backflow. Finally, the role of mechanical tension must be addressed through targeted resistance training; by stimulating the mechanoreceptors within the fascia, patients can promote TGF-β modulation, preventing the transition of fibroblasts into myofibroblasts, which is the primary driver of the irreversible "nodular" stage of Lipoedema. This multi-layered approach ensures that the oestrogen-adipose axis is not merely managed, but biologically re-engineered for resilience.
Summary: Key Takeaways
The oestrogen-adipose axis functions as a bidirectional regulatory network where fluctuating steroid concentrations dictate the structural integrity of subcutaneous adipose tissue (SAT). Evidence synthesised from *PubMed* and *The Lancet* confirms that Lipoedema is fundamentally a hormone-dependent microvascular and lymphatic failure, distinct from metabolic obesity. During the pubertal transition, a surge in 17β-oestradiol disproportionately activates oestrogen receptor alpha (ERα) in gynoid SAT deposits, inducing aberrant hyperplastic adipocyte expansion and pathological extracellular matrix (ECM) remodelling. At INNERSTANDIN, we identify this as the primary catalyst for interstitial fluid stasis and chronic tissue hypoxia.
Progression is further exacerbated during menopause; the decline in circulating oestrogen disrupts the ERα:ERβ ratio, triggering a shift toward systemic inflammation and densification of the adipose interstitium via TGF-β-mediated fibrosis. This hormonal volatility impairs lymphangiogenesis and increases microvascular permeability, resulting in the high-protein oedema characteristic of advanced stages. Within the UK clinical context, the oestrogen-adipose axis explains why these life-cycle milestones act as critical "tipping points" for lymphatic decompensation. The biological reality is a self-perpetuating cycle of adipocyte hypertrophy, hypoxia-inducible factor-1α (HIF-1α) activation, and subsequent lymphatic insufficiency, shattering the reductive fallacy that Lipoedema is a lifestyle-driven condition. This axis represents the nexus of endocrine signalling and lymphatic health, necessitating a paradigm shift in therapeutic intervention.
This article is provided for informational and educational purposes only. It does not constitute medical advice, clinical guidance, or a substitute for professional healthcare. Information reflects cited research at time of publication. Always consult a qualified healthcare professional before acting on any health information.
RESEARCH FOUNDATIONS
Biological Credibility Archive
Citations provided for educational reference. Verify via PubMed or institutional databases.
Medical Disclaimer
The information in this article is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your diet, lifestyle, or health regime. INNERSTANDIN presents alternative and research-based perspectives that may differ from mainstream medical consensus — these should be considered alongside, not instead of, professional medical guidance.
Read Full DisclaimerReady to learn more?
Continue your journey through our classified biological research.
DISCUSSION ROOM
Members of THE COLLECTIVE discussing "The Oestrogen-Adipose Axis: Why Puberty and Menopause Trigger Lipoedema Progression"
SILENT CHANNEL
Be the first to discuss this article. Your insight could help others understand these biological concepts deeper.
THE ARSENAL
Based on Lymphoedema — products curated by our research team for educational relevance and biological support.

Energy Blend Supports

Lugol’s Iodine – Hormonal Issues, Menopause, Immune System, Brain Fog, Memory, Thyroid, Dry Skin
INNERSTANDING may earn a commission on purchases made through these links. All products are selected based on rigorous educational relevance to our biological research.
RABBIT HOLE
Follow the biological thread deeper